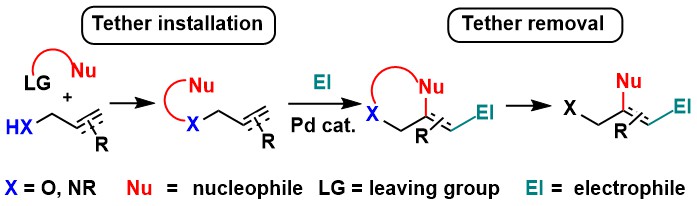
The tether approach
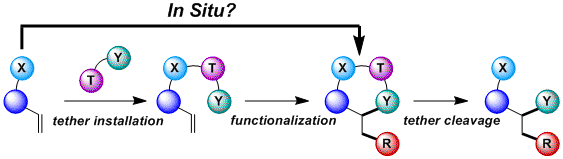
The intermolecular functionalization of olefins presents important challenges of reactivity, regio- and stereoselectivity. A classical solution to these issues is the installation of a tether making use of a functional group on the molecule. The now intramolecular reaction is faster and a cyclic transition state facilitates control over regio- and stereo- selectivity. Nevertheless, this comes at the cost of lower synthetic efficiency, as multiple steps are needed to install and remove the tether. Recently, researchers have begun to address this challenge by developing in situ tethering methods, making this strategy much more attractive (Chem. Sci. 2017, 8, 32. DOI: 10.1039/C6SC04366F ).
Trifluoroacetaldehyde-derived tethers for olefin functionalization
Since 2014, we had been attempting to develop an intermolecular version of the palladium-catalyzed oxy- and amino- alkynylation of olefins, but without success. In 2015, we turned to a new in situ-tethering approach based on the formation of half aminals. An efficient one-step procedure for the palladium-catalyzed oxy-alkynylation and arylation of allyl amines could be developed (Angew. Chem., Int. Ed. 2015, 54, 5250. DOI: 10.1002/anie.201500636 ). Key for success was the use of a hemiaminal tether derived from trifluoroacetaldehyde. The enhanced acidity of the half aminal allowed the use of a mild carbonate base.
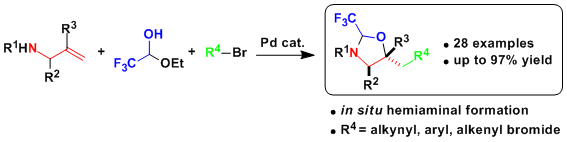
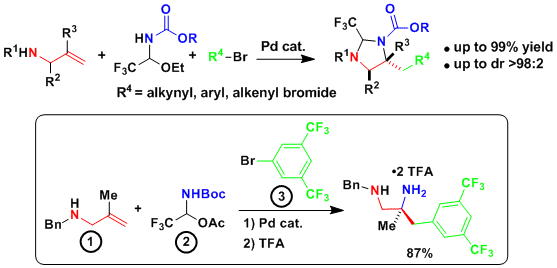
In 2016, we were able to extend this strategy to the synthesis of diamines via the in situ formation of aminals (Angew. Chem., Int. Ed. 2016, 55, 12881. DOI: 10.1002/anie.201607318 ). The reaction worked well for the introduction of different alkynes groups, as well as electron deficient (hetero)aryls. As the aminal tether and the amine protecting group can be easily removed under acidic conditions, the transformation constitutes a very efficient access to diamines.
In 2017, the strategy could be extended to allylic alcohols, resulting in aminoalcohols with different substitution patterns (Org. Lett. 2017, 19, 3548. DOI: 10.1021/acs.orglett.7b01524 ). In case of the aminoarylation reaction, it was necessary to develop a new Fu-XPhos ligand to suppress a competing Heck pathway.
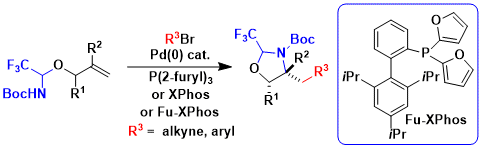
A limitation of the Pd0/II approach used in tethering was that it worked only for terminal alkenes and C-C bond formation. To overcome these limitations, we envisioned a PdII/IV pathway. In 2022, we were able to implement such a reaction for the aminoacetoxylation of styrene derived alkenes. Using simple PIDA as oxidant, aminodiols could be obtained after tether removal with good diastereoselectivity (Org. Lett. 2022, 24, 5068-5072. DOI: 10.1021/acs.orglett.2c01838 ).
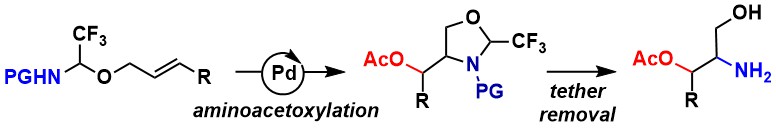
As an alternative approach to address the limitation of reactivity of substituted alkenes, we wondered if strained alkenes embedded in small rings, such as in cyclopropenes, could be functionalized under Pd0/II conditions. This would also realize an interesting connection with our research program on strained rings. In 2026, we successfully applied this strategy to the carboetherification of cyclopropenes using our trifluoroacetaldehyde-based tether on amines. This reaction gave access to highly substituted spirooxazolidine products with perfect diastereoselectivity (Chem. Sci. 2026, 17, in press. DOI: 10.1039/D5SC09351A ).
Functionalization of alkynes
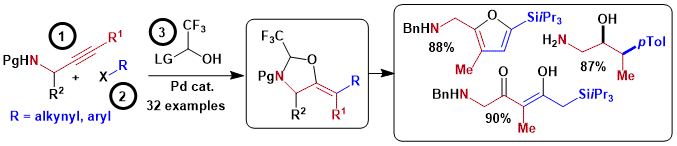
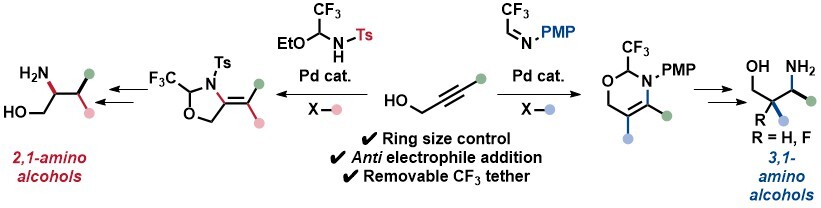
We then investigated if the tethering approach could be extended to alkynes. We were pleased to see that the carbooxygenation reaction was successful under similar conditions. The use of alkynes had several advantages: the reaction was also successful for substituents at the terminal position, and a broader range of aryl groups was tolerated as cross-coupling partners. Interestingly, the reaction proceeded with cis selectivity for terminal alkynes, but trans selectivity for internal ones. The products could be easily transformed into furan derivatives, or reduced with high diastereoselectivity (Chem. Eur. J. 2019, 25, 3010-3013. (DOI:10.1002/chem.201900020 ). It was also possible to access oxazolidinone products by using carbonate salts as carbon dioxide surrogates. (Eur. J. Org. Chem. 2019, 2019, 5183-5186. DOI:10.1002/ejoc.201900500 )
We then investigated propargylic alcohols as starting materials, with the aim to access enamines stereoselectively through a tethered carboamination reaction. Interestingly, the reaction had a different outcome depending on the protecting group on the nitrogen: a 5-exo cyclization leading ultimately to 1,2-amino alcohols was observed with a tosyl group, whereas 6-endo cyclization giving 1,3 amino alcohols was obtained with a PMP group. In the case of the 6-membered heterocycles, an efficient fluorination method could be used to access fluorinated amino alcohols with high diastereoselectivity. (Angew. Chem., Int. Ed. 2024, 63, e202411383. (10.1002/anie.202411383).
Enantioselective Transformations
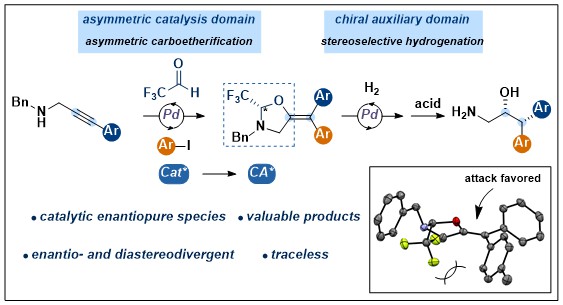
We found the perfect diastereoselectivity observed for the hydrogenation of the alkyne carbooxygenation products fascinating: Indeed, if the N,O-acetal could be installed enantioselectively, a highly stereoselective synthesis of difficult to access amino alcohols would result. Conceptually, this would constitute the “catalytic formation of a chiral auxiliary”, an approach mostly unexplored in synthetic chemistry. It would have the potential to combine the robustness and high selectivity obtained with chiral auxiliaries with the higher atom- and “chiral element” economy of enantioselective catalysis. As we knew that formation of the initial half aminal was reversible, a dynamic kinetic asymmetric transformation with a chiral palladium catalyst appeared possible. Realisation of this idea took two years, as identifying the best ligand was especially challenging. In 2020, we finally succeed, based on the use of a truncated “one-arm” Trost-type ligand. High enantioselectivity was observed in the carboxygenation step, and again perfect diastereoselectivity in hydrogenation. Deprotection was easy to give the free amino alcohols (J. Am. Chem. Soc. 2020, 142, 17334-17339. DOI:10.1021/jacs.0c09177 ).
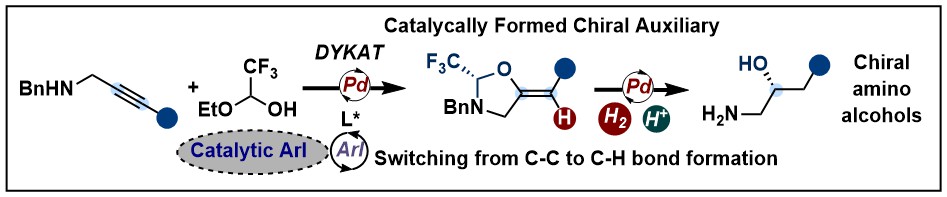
A limitation of the catalytically formed auxiliary approach was the low yield and enantioselectivity observed with unsubstituted alkynes. Therefore, monoarylated amino alcohols could not be accessed efficiently. In 2022, we could turn a side reaction -the protodemetallation- into the main process to achieve a highly enantioselective trans-hydroalkoxylation of propargylic amines. A surprising result of this research was the counterintuitive use of an aryl iodide additive to enhance both the yield and enantioselectivity of the desired transformation. (ACS Catal. 2022, 12, 7565-7570. DOI:10.1021/acscatal.2c01809 ).
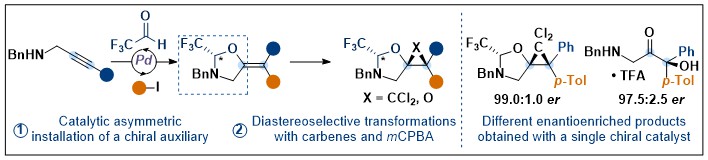
As a next step, it was important to demonstrate the generality of the catalytically formed auxiliary concept beyond hydrogenation. In 2022, we showed that the concept was also successful in the case of C-C and C-O bond formation. We were able to develop both an asymmetric cyclopropanation and asymmetric epoxidation proceeding with good diastereoselectivity with the formed auxiliary (Angew. Chem., Int. Ed. 2022, 61, e202113925. DOI:10.1002/anie.202113925 ).
In 2022, we also reviewed the recent progress in the field of Pd-catalyzed functionalization of alkenes and alkynes based on the used of removable tethers (Tetrahedron 2022, 128, 133135. DOI:10.1016/j.tet.2022.133135).