Acetylenes
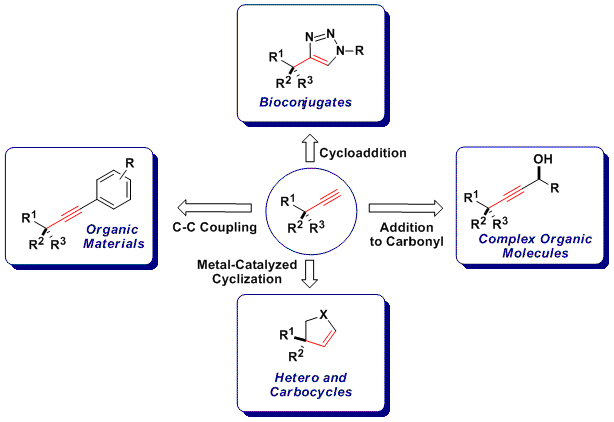
Alkynes are versatile functionalities in organic synthesis, as they participate for example in addition reactions to carbonyl compounds, as well as cycloaddition and coupling reactions. These processes have found widespread applications in neighbouring fields, such as biochemistry and material sciences. There is consequentyl an urgent need for new methods to access acetylenes efficiently, especially when considering that research in alkynylation reactions has been much less intensive than for alkenylation or arylation methods.
Nucleophilic vs electrophilic alkynylation
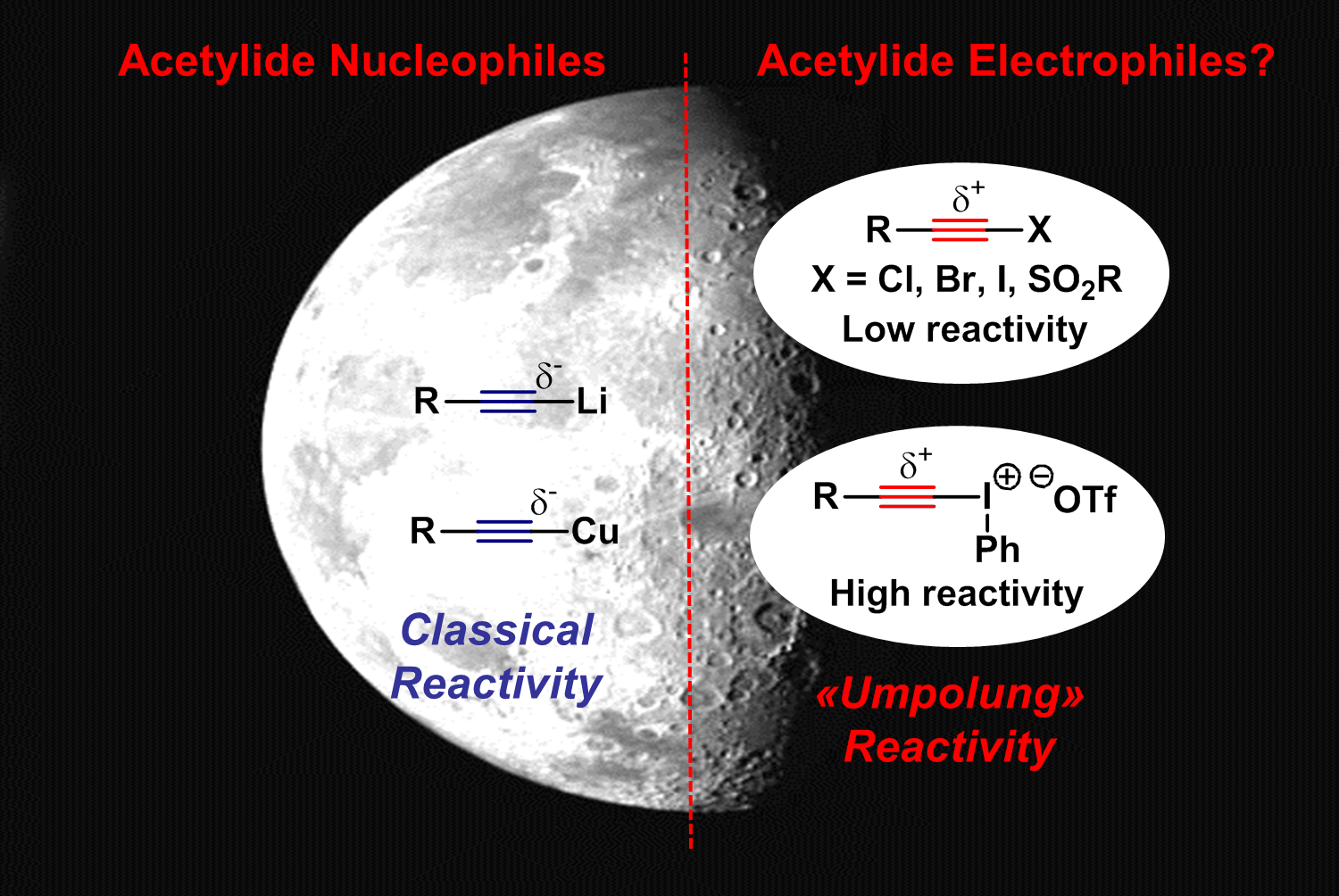
The classical synthon to transfer an alkyne to an organic molecule is an acetylide anion, which can be easily accessed by deprotonation of a terminal acetylene. On the other hand, the availability of electrophilic acetylene synthons would be highly useful to increase the flexibility of alkyne synthesis. The most obvious choice would be acetylene halides, but they present in many cases a too limited reactivity. On the other hand, hypervalent iodine reagents are much stronger electrophiles, due to the very weak hypervalent bond. We consequently decided to explore this “dark side” of acetylene chemistry and to make use of this exceptional reactivity for the development of new catalytic electrophilic alkynylation methods (Chem. Soc. Rev. 2012, 41, 4165-4179. DOI:10.1039/C2CS35034C).
Hypervalent iodine: Heterocyclic alkynylation reagents
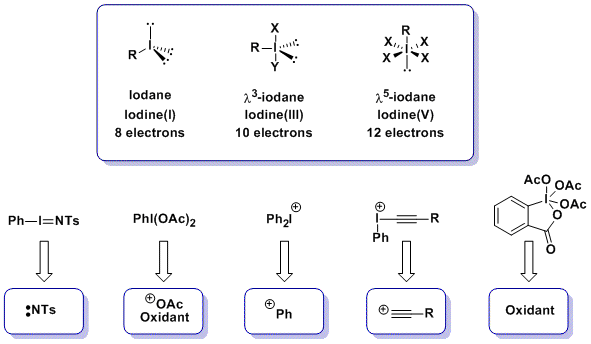
The non-classical 4-electrons 3 centres bonds of hypervalent iodine are weaker than normal classical bonds. This confers an exceptional reactivity to these compounds as oxidants or atom-transfer reagents. The reactivity of organic synthons can be inverted (Umpolung) and electrophilic reagents based on carbon, nitrogen or oxygen can be obtained. In the case of electrophilic carbon-transfer, much work has focused on aryl transfer and the potential of acetylene transfer, especially in catalytic reactions, was mostly unexplored in 2008.
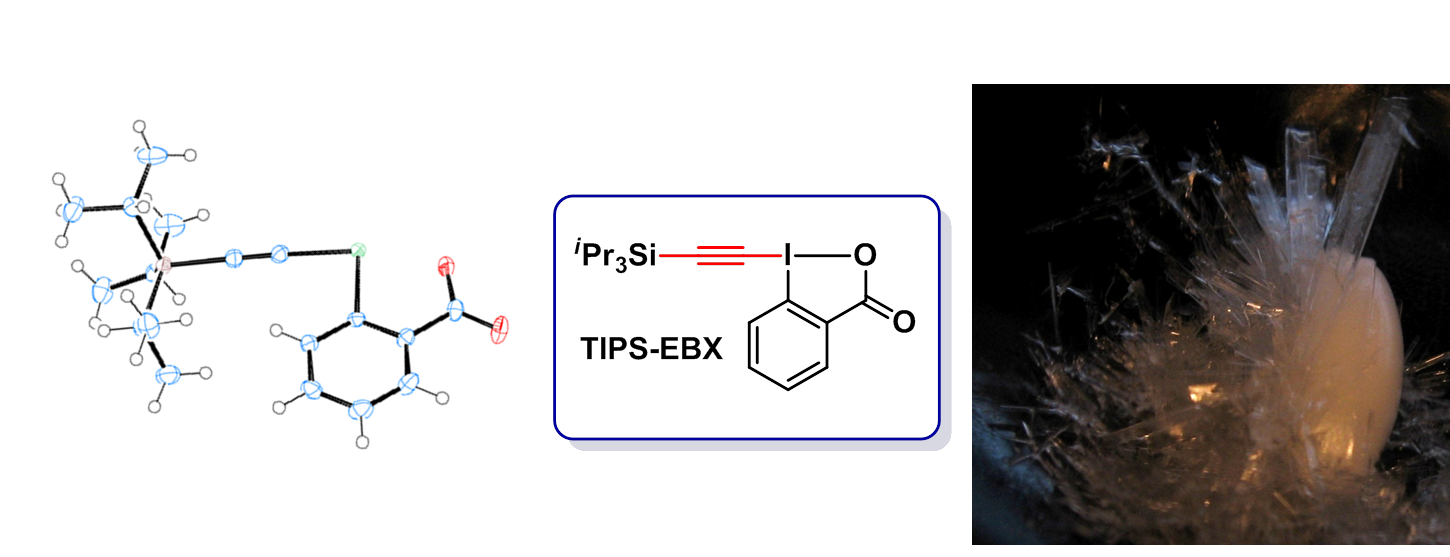
However, be became quickly aware that classical alkynyliodonium salts were not compatible with metal catalysis. We consequently turned to EthynylBenziodoXolone (EBX) reagents (Chem. Commun. 2011, 47, 102. DOI:10.1039/C0CC02265) introduced by Ochiai and Zhdankin, which had never been used in acetylene transfer reactions. In particular, the triisopropylsilyl protected reagent (TIPS-EBX) was very successful and is now commercially available from Sigma-Aldrich (Synthesis 2012, 44, 1155. DOI: 10.1055/s-0031-1290589). In 2019, we developed a one-step synthesis of TIPS-EBX in collaboration with Merck (Org. Proc. Res. Dev. 2020, 24, 106. DOI: 10.1021/acs.oprd.9b00498). Over the year, diverse reagents were synthesized, including modified ethynylbenziodoxolones (EBXs), the alcohol-derived ethynylbenziodoxoles (EBxs) and the nitrogen-derived ethynylbenziodazol(on)es (EBZs, EBzs).
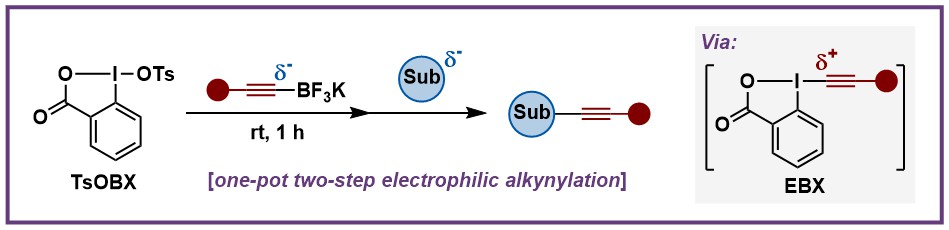
The syntheses of EBX reagents developed by our group, as well as the Ochiai, Zhdankin and Olofsson groups, are highly efficient. Nevertheless, they are still drawbacks, such as sensitive starting materials in the case of alkynyl boronic esters or low yields in case of silyl alkynes, especially for alkyl substituted EBX synthesis. All the approaches so far required purification of the reagents before using them in further applications. In 2021, we developed a “perfect” synthesis of EBX reagents from alkynyl trifluoroborate salts and tosyloxybenziodoxolone, requiring no Lewis acid activator and giving the EBX in >95% yield and >95% purity. This enabled us to use them directly in one-pot processes, giving a fast access to diversified products without the time-consuming and expensive isolation and purification steps required previously (Org. Lett. 2022, 24, 142. 10.1021/acs.orglett.1c03771).
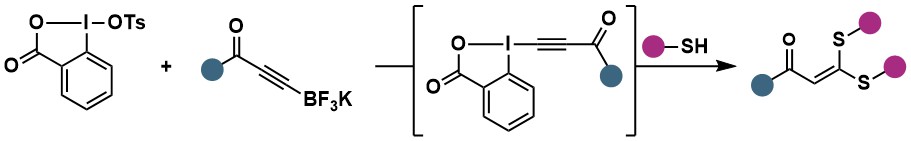
In 2023, we used this new synthesis method to access highly electrophilic and unstable Acyl-EBX reagents in situ (Org. Lett. 2023, 25, 7535-7539. 10.1021/acs.orglett.3c02869). The reagents were then applied to the synthesis of ketene dithioarylacetals via double addition of thiol nucleophiles.
2-Iodobenzoic acid derivatives are by far the most useful EBX reagents. Nevertheless, we and others found over the year that more stable bistrifluoromethyl EBx derivatives were superior in some case (vide infra). Nevertheless, the use of these reagents was impaired during several years due to their inefficient multi-step synthesis. In 2023, we adapted the Olofsson protocol for the efficient one-step synthesis of these reagents (Chem. Commun. 2023, 59, 12637-12640. 10.1039/D3CC04525K).
As the number and structural diversity of EBX, EBX and EBZ reagents were increasing rapidly, we collected all their NMR and X-ray structural data in an overview article in 2023 (Helv. Chim. Acta 2023, e202200175. (DOI: 10.1002/hlca.202200175 ).
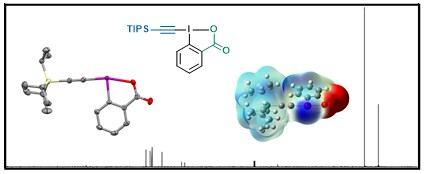
Synthesis of alkynylated aromatic compounds
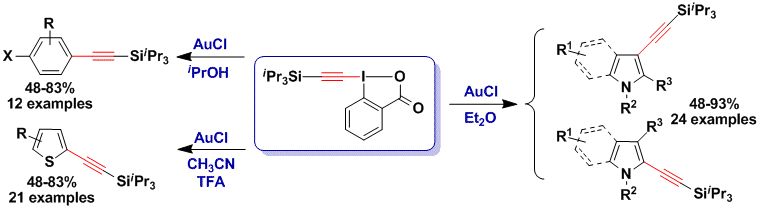
Using TIPS-EBX and AuCl as catalyst, the direct alkynylation of indoles and pyrroles became possible at room temperature in open flask (Angew. Chem., Int. Ed. 2009, 48, 9346. DOI: 10.1002/anie.200905419). Interestingly, this reaction can also be applied to the alkynylation of tryptophan in peptides (Beilstein J. Org. Chem. 2016, 12, 745. DOI: 10.3762/bjoc.12.74). The reaction could be later extended to anilines (Org. Lett. 2012, 14, 744. DOI: 10.1021/ol203289v). Finally, a cooperative effect between gold and Bronsted acids was discovered for the alkynylation of thiophenes (Angew. Chem., Int. Ed. 2010, 49, 7304. DOI: 10.1002/anie.201003179).
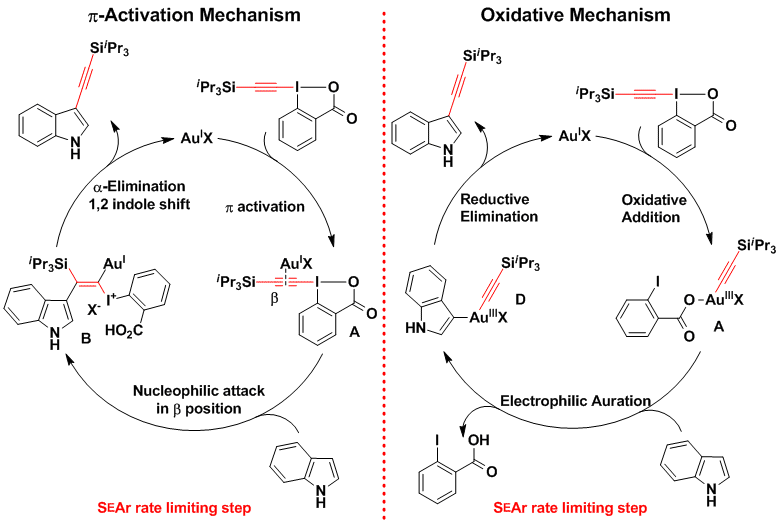
Several investigations involving reaction kinetics, isotope labeling and structure activity relationship of the EBX reagents are in agreement with two alternative mechanisms: pi-activation and oxidative addition involving Au(III) intermediates (Chem. Eur. J. 2012, 18, 5655. (DOI:10.1002/chem.201200200 ).
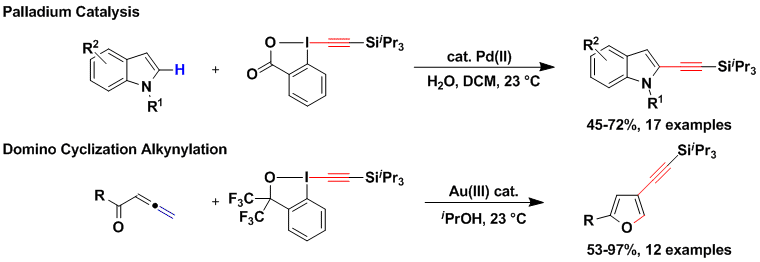
The developed gold-catalyzed alkynylation usually displayed high regioselectivity for functionalization of the most electron-rich position of aromatic compounds. Consequently, it cannot be used if compounds alkynylated at other positions are desired. Two strategies were followed to overcome this limitation. First, the use of a palladium catalyst in the case of indoles allowed to shift the alkynylation regioselectivity from C3 to C2 (Org. Lett. 2013, 15, 112. (DOI:10.1021/ol3031389 ). Second, a completely different approach was used to access C3-alkynylated furans via a domino process combining the cyclization of ketoallenes developed by Hashmi and co-workers with an unprecedented alkynylation step (Angew. Chem., Int. Ed. 2013, 52, 6743. (DOI:10.1002/anie.201302210 ). Interestingly, fine-tuning of the gold catalyst and modification of the hypervalent iodine reagent were essential for an efficient reaction. For several years, we were puzzled by the fact that this reaction was catalyzed by a Au(III) catalyst. In 2017, calculations obtained in collaboration with the computational chemistry group of Prof. Alireza Ariafard suggested that the Au(III) catalyst was in fact reduced in situ to Au(I), which was the true active catalyst. In fact, we obtained the same result using a Au(I) picolinate catalyst. The picolinate ligand was essential to lower the energy of the key oxidative addition transition state (Dalton Trans. 2017, 46, 12257. ( DOI:10.1039/C7DT03154H ).
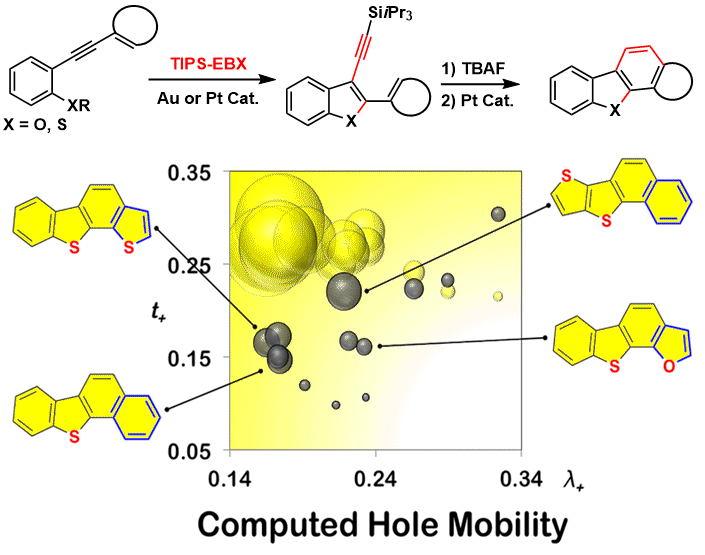
In 2017, the domino cyclization-alkynylation reaction was used in a new approach towards heterotetracenes (Chem. Eur. J. 2017, 23, 8058. (DOI:10.1002/chem.201701139). Heterotetracenes are important building block for organic materials. In fact, calculations performed by the Corminboeuf group indicated that the new tetracenes should have very high ion mobility and should be interesting for applications. Unfortunately, attempts with the Sivula group to generate thin films with these molecules did not result in good performance, given often pretty, but random crystallization.

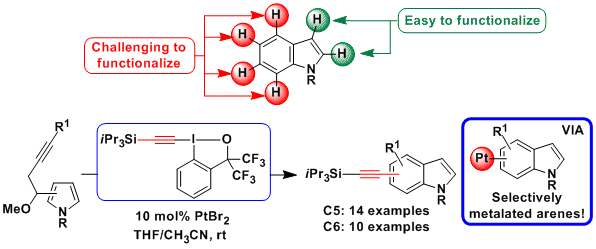
Up to 2015, a major limitation of the domino reactions was that they could be applied only to the synthesis of alkynylated heterocycles. Access to indoles alkynylated on the benzene ring instead would be highly interesting, as it is not possible to access these compounds via C-H functionalization when the more reactive C2/C3 positions are unsubstituted. In 2015, a new domino process based on the platinum-catalyzed nucleophilic attack of a pyrrole ring onto an alkyne followed by alkynylation with EBX reagents was developed (Angew. Chem., Int. Ed. 2015, 54, 5438. (DOI:10.1002/anie.201412321 ). Both C5- and C6- alkynylated indoles could be accessed selectively.
Catalytic oxy and amino alkynylation of olefins
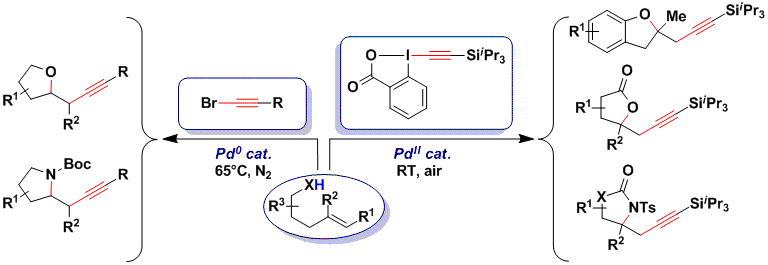
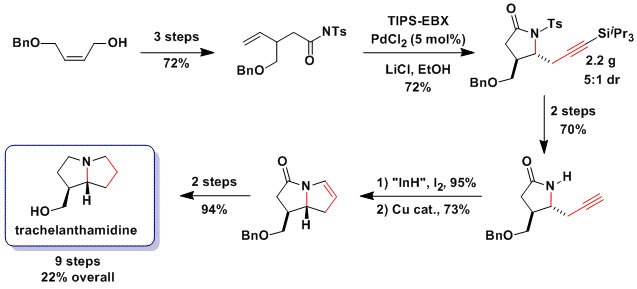
The multi-functionalization of olefin ussing Pd catalysts is an intensive field of research, as such transformations allow a very fast access into molecular complexity starting from bulk chemicals. Nevertheless, oxy- or amino- alkynylations of olefins were unknown before our work. We used the exceptional properties of TIPS-EBX in the Pd (II)-catalyzed intramolecular oxy- and amino- alkynylation of olefins for the synthesis of lactones (Org. Lett. 2010, 12, 384. DOI: 10.1021/ol9027286 ) and lactams (Angew. Chem., Int. Ed. 2011, 50, 4680. DOI: 10.1002/anie.201100718 ) at room temperature in open flasks. The utility of the reaction was then demonstrated in the total synthesis of the small pyrrolizidine alkaloid trachelanthamidine. Unfortunately, the method could not be used for the synthesis of pyrrolidines and tetrahydrofurans. In this case, using an easier to reduce Pd(0) phosphine complex together with a bromoacetylene as a weaker oxidant was successful (Org. Lett. 2011, 13, 6324. (DOI: 10.1021/ol2029383 ). The main advantages of this new protocol is that it also work for more complex aliphatic alkynes and can be used for C-C bond formation at secondary positions (J. Org. Chem. 2013, 78, 3783. (DOI:10.1021/jo400254q ).
Copper-catalyzed alkynylations
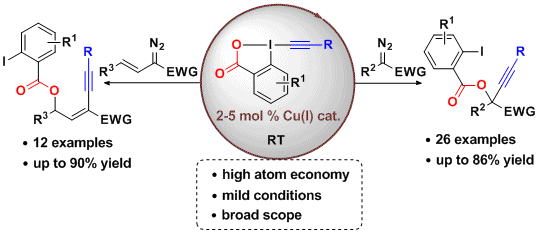
Up to 2015, a major disadvantage of reactions involving EBX reagents was that a stoichiometric amount of 2-iodobenzoic acid was formed as a waste. We therefore attempted to also use this part of the reagent in the transformation. In 2016, were reported the first oxyalkynylation of diazo compounds, in which both the alkyne and the iodobenzoate are incorporated into the formed product (J. Am. Chem. Soc. 2016, 138, 2190. DOI: 10.1021/jacs.6b00278). This first highly atom-economical transformation of EBX reagents is also remarkable for its broad scope of both alkynes and diazo compounds. It is also one of the rare examples of the use of a copper catalyst with EBX reagents. In the case of vinyldiazo compounds, conjugated enynes were obtained as the only products.
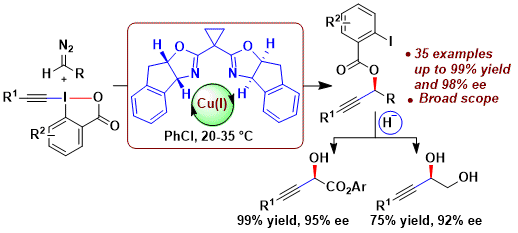
In 2017, a further breakthrough was achieved in this area: the first enantioselective oxyalkynylation of diazo compounds. Using a commercially available BOX ligand, high enantioselectivities were obtained, especially when diazo compounds derived from bulky aryl esters were used. (J. Am. Chem. Soc. 2017, 139, 8420. DOI: 10.1021/jacs.7b04756). The obtained products could be easily reduced to useful propargylic alcohols or diols without loss of enantiopurity.
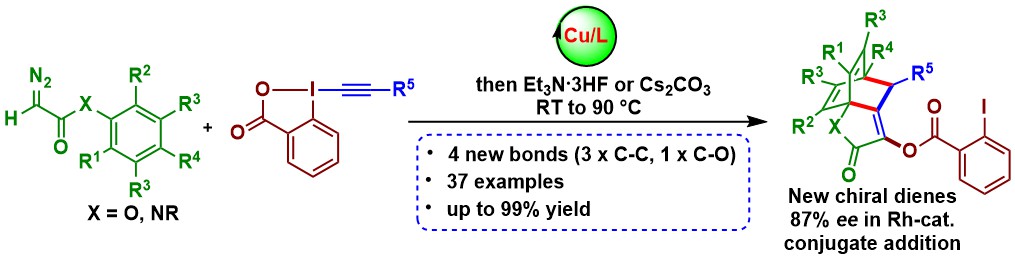
Serendipity remains an essential factor in research in organic chemistry. This was again demonstrated when we attempted deprotection of the silylated alkynes obtained through oxyalkynylation. A very uncommon [4+2] cycloaddition between the formed allene and the benzene ring on the ester group was observed – a so called Himbert reaction. We were able to optimized a one-pot oxyalkynylation-cycloaddition process. (Angew. Chem., Int. Ed. 2021, 60, 5475-5481. DOI: 10.1002/anie.202012299). The obtained products could be directly used as chiral ligands for the enantioselective rhodium-catalyzed conjugate addition to cyclohexenone.
Accessing amino acid derivatives using this methodology would be highly attractive. In 2018-2019, we made numerous attempts towards this goal. Our first approach was to develop a new alkynylation reagents based on the benziodazolone (BZ) heterocycle. The new EBZ reagents could indeed be accessed in high yield. However, despite the presence of the nitrogen atom bound to iodine, they still acted as oxygen-transfer reagents. Nevertheless, very high enantioselectivities could be achieved with simpler alkyl diazo esters as substrates (Chem. Eur. J. 2019, 25, 9522-9528. DOI:10.1002/chem.201900950 ).
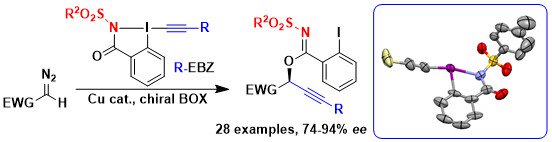
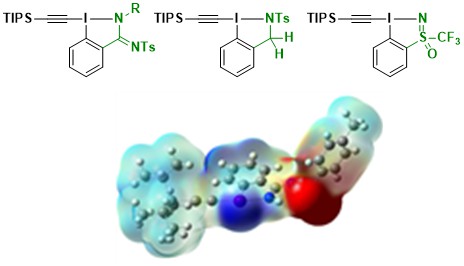
The next logical attempt towards amino acid synthesis was preparing reagents lacking the nucleophilic amide oxygen atom. We successfully synthesized new amidine- and sulfonamide-based reagents, as well as sulfoximine-based ones together with the Magnier group at the Institut Lavoisier de Versailles. However, these new reagents did not react anymore with diazo compounds (Chem. Eur. J. 2021, 27, 10979-10986. 10.1002/chem.202101475</a).
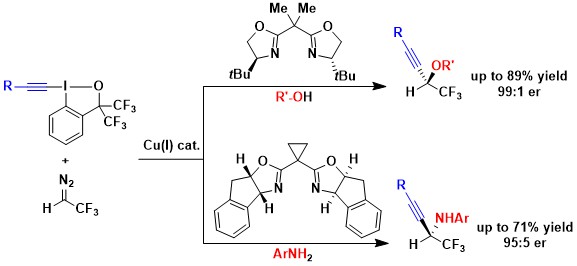
From the point of view of atom economy, the reaction between diazo compounds and EBX/EBZ reagents we developed were interesting, but they were limited concerning product diversity. In this respect, the incorporation of an external electrophile different from 2-iodo benzoate would be highly interesting. In 2020, we finally achieved a breakthrough with the first three component reaction of EBX reagents, alcohols and diazo compounds. (Chem. Eur. J. 2020, 45, 10199-10204. DOI:10.1002/chem.202001317 ). Key for success was the use of the bis-trifluoromethyl derived benziodoxole reagent instead of benziodoxolones.
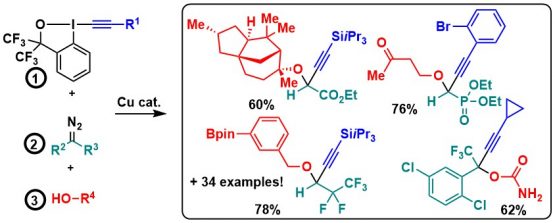
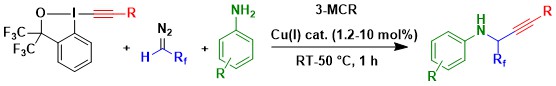
In 2021, we finally found a solution for the addition of nitrogen nucleophiles by developing a three component reaction between fluorinated diazo compounds, anilines and alkynyl benziodoxole reagents. This was an important breakthrough, but the scope of the reaction remained narrow: ester-substituted diazo compounds did not work, and only anilines could be used as nucleophiles (J. Org. Chem. 2021, 86, 10928-10938. 10.1021/acs.joc.1c01423).
The development of enantioselective three component reactions was especially challenging. In 2023, we finally succeeded in the case of fluorinated diazo compounds. Using a simple copper BOX catalyst, the three component reaction of both alcohols and anilines was successful (Angew. Chem., Int. Ed. 2023, 62, e202305776. DOI: 10.1002/anie.202305776). This transformation displayed a strong catalyst control, enabling highly diastereoselective reactions in case of chiral substrates.
In general, combining the reactivity of carbenes and carbenoids and cyclic hypervalent iodine reagents has led to the development of many new transformations in the last decade. This motivated us to write a review on the topic in 2024 (Synthesis 2024, 56, 3815-3828. DOI: 10.1055/a-2418-8285 ).

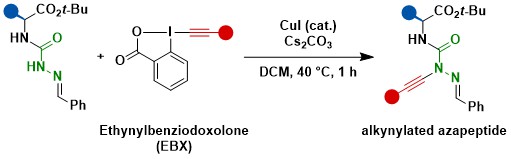
In 2022, when attempting the in-situ formation of non-stable diazo compounds from hydrazones, we discovered an efficient copper catalyzed C-N alkynylation. This transformation could be used for the synthesis of alkynylated azapeptides in high yields (Org. Lett. 2022, 24, 6614. DOI: 10.1021/acs.orglett.2c02625).
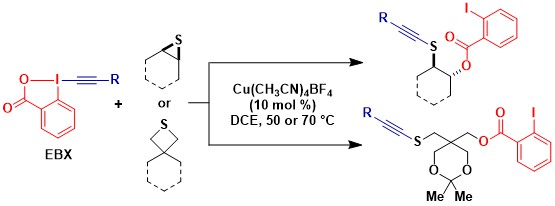
In 2019-2020, we also discovered a second atom-economical copper-catalyzed oxyalkynylation involving thiiranes and thiethanes. (Org. Lett. 2020, 22, 422-427. DOI:10.1021/acs.orglett.9b04157). Both three- and four-membered rings reacted to give the corresponding thioalkynes with simultaneous attack of 2-iodobenzoate.
Alkynylation of nucleophiles without transition metals
The exceptional combination of reactivity and stability characterizing the EBX reagents is not only useful in the case of metal catalysis, but also lead to important successes in the metal-free alkynylation of nucleophiles. Removal of the silyl group in situ from the corresponding reagent TMS-EBX gives the extremely reactive EBX reagent, which is able to alkynylate stabilized enolates at -78 °C (Chem. Eur. J. 2010, 16, 9457. DOI: 10.1002/chem.201001539). The reaction was successful for keto, nitro and cyano esters and sets the bases for the synthesis of non-natural alkynylated amino acids. Under phase-transfer conditions using Maruoka’s binaphthyl-derived catalyst, the first example of asymmetric induction with up to 79% ee could be achieved (Adv. Synth. Catal. 2013, 355, 1631. DOI: 10.1002/adsc.201300266). During this work, the first safety profiling of EBX reagents was also done in collaboration with Syngenta Crops Science.
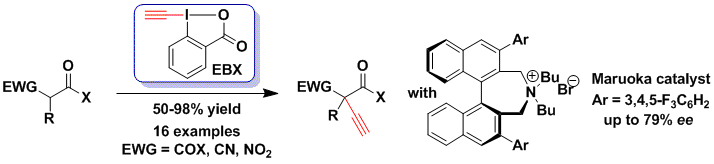
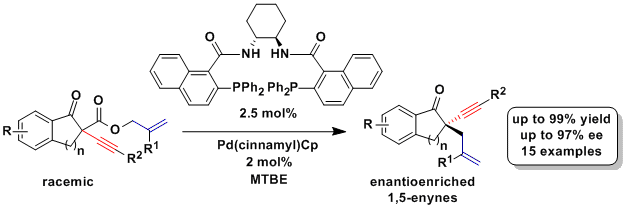
Although the first example of asymmetric alkynylation of ketoesters represented an important breakthrough, the obtained asymmetric induction remained moderate. In order to obtain a more efficient synthesis of enantioenriched all-carbons propargylic centers, we turned to a dynamic asymmetric transformation using easily accessible racemic ketoesters: the palladium-catalyzed decarboxylative allylation, first discovered by Tsuji and later made asymmetric by Stoltz and Trost. Indeed, this approach proved highly successful when using the Trost cyclohexyldiamine-naphthyl-based ligand and we were now able to access for the first time propargylic quaternary centers with enantiomeric excess up to 97% (Org. Lett. 2014, 16, 5768. DOI: 110.1021/ol5028333). The 1,5-enynes obtained using this method are highly useful building blocks which could be transformed into different polycyclic carbocycles using metathesis or gold and platinum catalysis.
The main focus of our work has been in the alkynylation of C-nucleophiles to construct the carbon backbones of organic molecules. The alkynylation of heteroatoms such as nitrogen, oxygen or sulfur leads to alkynes with high nucleophilicity, which are highly useful as building blocks in synthetic chemistry. Whereas efficient methods have emerged for the alkynylation of nitrogen compounds to give ynamides, ynol ethers and thioalkynes are still very difficult to access. Using TIPS-EBX in presence of tetramethylguanidine as base, a highly practical and efficient alkynylation of thiols was developed (J. Am. Chem. Soc. 2013, 135, 9620. DOI: 10.1021/ja4044196). The reaction proceeded at room temperature in an open flask and was usually finished in less than one minute. It was successful in the case of aliphatic, aromatic, heteroaromatic and peptidic thiols. For example, the selective alkynylation of cysteine in a dipeptide could be achieved even in the presence of the unprotected lysine and histidine amino acids. Cycloaddition of the desilylated thioalkyne with an azide allowed the efficient installation of a fluorophore.
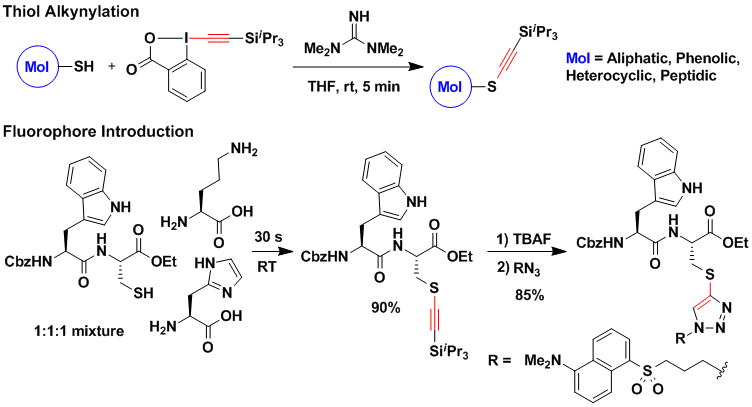
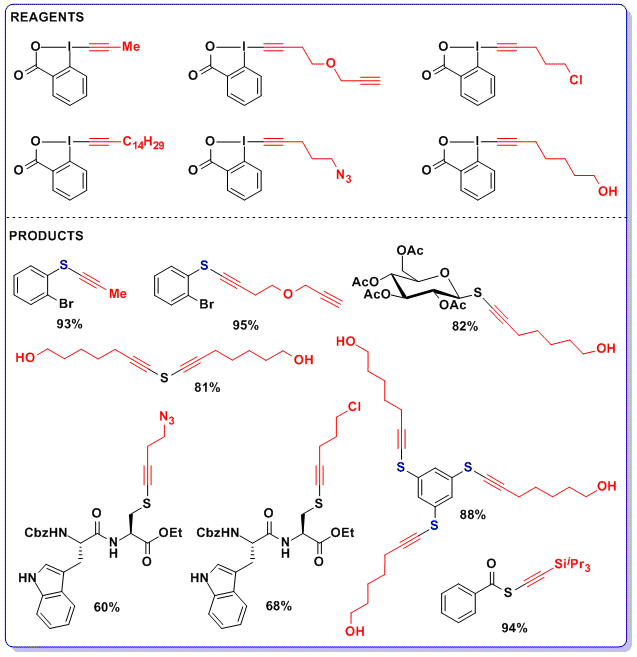
One major limitation of the alkynylation reactions so far had been the requirement for bulky silyl groups on the alkyne. In 2014, we discovered that the thiolalkynylation reaction had a much broader scope and was also very efficient in the case of aryl and alkyl substituents (J. Am. Chem. Soc. 2014, 136, 16563. DOI: 10.1021/ja5083014). The advantage of the enhanced stability of benziodoxolones became apparent again, as we were now able to synthesize reagents containing functional groups such as alkynes, halogens, azides or alcohols. Alkyne transfer to a broad range of thiols including thioacids, peptides, glycosides, (poly)thiophenols and simple sodium hydrogen sulfide was then successful.
The mild conditions, fast rate and functional group tolerance of the thiol alkynylation reaction motivated us to start our research program on the modification of biomolecules.
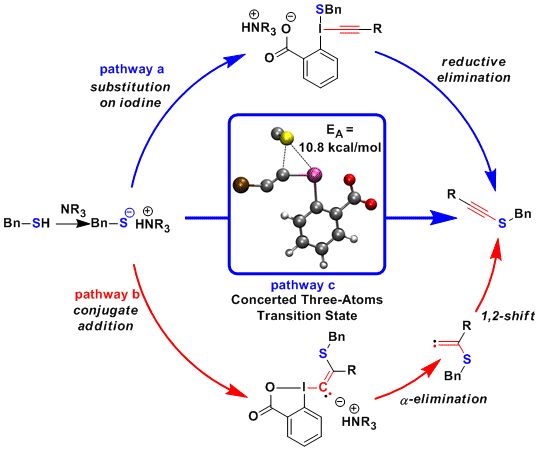
The exceptional efficiency and generality of the thiol alkynylation rose the question of the reaction mechanism. In principle, most hypervalent iodine mediated reactions have been proposed to proceed via an addition-elimination mechanism on iodine (pathway a). Nevertheless, for alkynyliodonium salts, a conjugated addition, alpha-elimination, 1,2-shift mechanism is usually favored (pathway b). Indeed, small amounts of side products resulting from this pathway have been isolated for Me-EBX. To further understand these results, we turn to computational chemistry (J. Am. Chem. Soc. 2014, 136, 16563. DOI: 10.1021/ja5083014). The computations confirmed that pathway b was energetically feasible. In contrast, no addition intermediate on iodine could be computed. Instead, a new concerted low energy mechanism was identified, proceeding via a three-atom transition state (pathway c). This pathway was found to be favored with silyl substituents, whereas both pathways c and b were closer in energy with alkyl substituents.
In 2015, a further mechanism could be finally identified via calculations: Initiated by a iodine sulfur interaction like for the concerted pathway, a shift of the sulfur towards the beta carbon finally results in conjugate addition (Org. Lett. 2016, 18, 60. DOI: 10.1021/acs.orglett.5b03241). This pathway was favored for electron-donating groups on the alkyne, such as alkyls, whereas both the concerted and conjugate addition pathway were possible for silyl and aryl substituents.
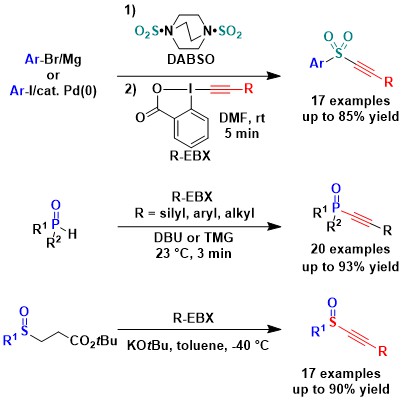
The alkynylation reaction could be also extended to sulfite salts. A convenient one-pot procedure starting from aryl iodide or Grignard reagents using DABSO and EBX reagents was developed (Org. Lett. 2015, 17, 736. DOI: 10.1021/acs.orglett.5b00015 ). In addition, we discovered that the alkynylation of H-phospi(na)tes and secondary phosphine oxides was also possible with silyl, aryl and alkyl EBX reagents (Chem. Commun. 2014, 50, 12923. DOI: 10.1039/C4CC06851C ). In 2019, we extended the alkynylation reaction to in situ generated sulfinate salts. (J. Org. Chem. 2019, 84, 3687-3701. DOI:10.1021/acs.joc.9b00050 )
Alkynylation via photoredox catalysis
Recently, EBX reagents have been demonstrated to be excellent reagents for the alkynylation of alkyl radicals. Photoredox catalysis has emerged as one of the mildest methods to generate radicals, using simple visible light to promote the reaction. The decarboxylative alkynylation of aliphatic carboxylic acids could be achieved in high yields using an iridium photocatalyst and EBX reagents (Angew. Chem., Int. Ed. 2015, 54, 11200. DOI: 10.1002/anie.201505111). Silyl, aryl and alkyl alkynes could be accessed on tertiary, secondary and primary positions using this method. The reaction was especially efficient for amino acid derivatives. The Xiao group published a similar transformation simultaneously with our group (Angew. Chem., Int. Ed. 2015, 54, 11196. DOI: 10.1002/anie.201504559). The reaction was usually run with blue LED’s as light source, but sun light could also be used. This success with simple amino acids motivated us to start a research program on the modification of more complex peptides (see: Modification of Peptides and Proteins).


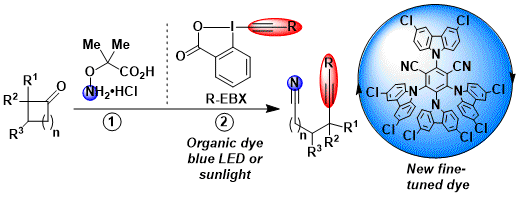
In 2018, we used a different approach for the generation of the radical: a fragmentation/alkynylation cascade of cyclobutyl oxime ethers led to the formation of alkynyl nitriles in good yield. The design of a new organic catalyst based on the CzIPN scaffold (the “dragoon catalyst” in our groups) was required to achieve high yields. The oxime ether can be synthesized directly in situ prior to the reaction by condensation of the ketone with the corresponding hydroxylamine. Calculation performed by the Corminboeuf group allowed us to better understand the properties of the dye. (Chem. Sci. 2018, 9, 5883-5889. DOI:10.1039/C8SC01818A )

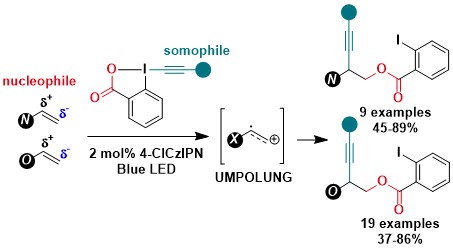
In 2020, a collaboration with the Kokotos group resulted in new methods for the C-H or deconstructive alkynylation of saturated heterocycles (Chem. Eur. J. 2020, 26, 14453-14460. DOI:10.1002/chem.202002868). Our first atom-economical reaction of EBX reagents under photoredox conditions was also developed in 2020: Based on the oxidation of electron-rich heteroatom-substituted alkenes to the corresponding radical cations by an organic dye, we were able to add both the alkyne and the iodobenzoate part of the reagent onto the double bond (Chem. Sci. 2020, 11, 11274-11279. DOI:10.1039/D0SC03655B).
In 2021, we were working on a new deoxygenative alkynylation of alcohols via cesium oxalate salts when me made a surprising discovery: An efficient background reaction was observed in absence of a photocatalyst in the case of aryl substituted EBX reagents. We realized that direct photoexcitation of ArEBXs was possible using strong Kessil lamps, and the excited hypervalent iodine reagent was a strong oxidant (+1.8 V). This allowed us to perform several alkynylations under oxidative conditions starting from oxalates, carboxylates, trifluoroborate salts or even C-H bonds without the need for a photoredox catalyst (Angew. Chem., Int. Ed. 2021, 60, 23287-23834. 10.1002/anie.202110257).
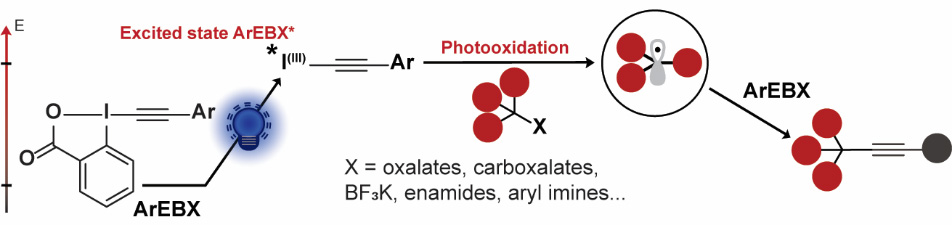
The development of other deoxygenation reactions was less successful, nevertheless we were able to obtain preliminary results for a deoxycyanation as well as for a desulfurative alkynylation (Helv. Chim. Acta 2022, e202200161. 10.1002/hlca.202200161).
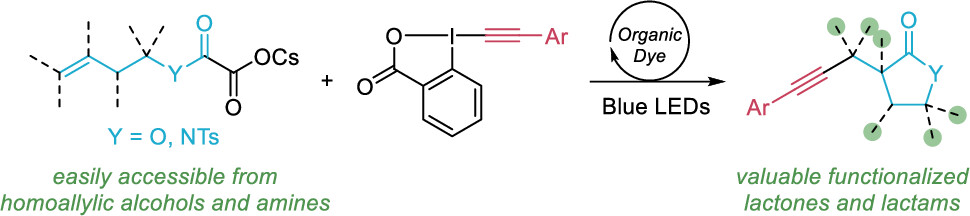
In 2024, we then further extended our photocatalyzed alkynylation reactions to a cyclization-alkynylation cascade giving access to important lactones and lactams starting from cesium oxalates (Org. Lett. 2024, 26, 4235-4239. 10.1021/acs.orglett.4c01078).
Alkynylation with EBX reagents: Now a matured field
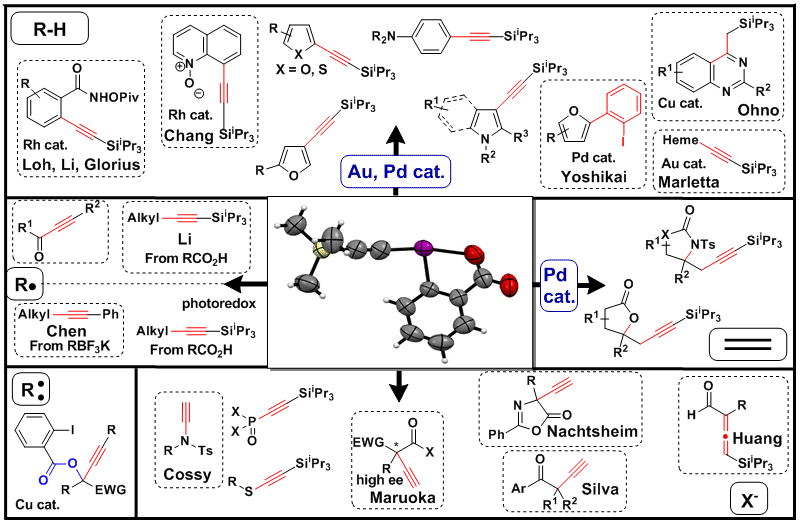
Since our discovery of the exceptional properties of EBX compounds for electrophilic alkynylation, many other groups have joined us in using these fascinating reagents in new transformations (Topp. Curr. Chem. 2015, 1-36. DOI: 10.1007/128_2015_660 and PATAI’S Chemistry of Functional Groups 2018, John Wiley & Sons, Ltd. DOI: 10.1002/9780470682531.pat095). A schematic overview of selected examples can be found in the figure below. In 2023, we wrote an updated review about this area of research. (Chem. Commun. 2023, 59, 1589-1604. DOI: 10.1039/D2CC06168F ). Finally, our last update about this area appeared as a chapter in Science of Synthesis in the volume dedicated to hypervalent iodine chemistry (Science of Synthesis Hypervalent Halogens in Organic Synthesis 2025, Ch. 6. DOI: 10.1055/sos-SD-246-00076 ).