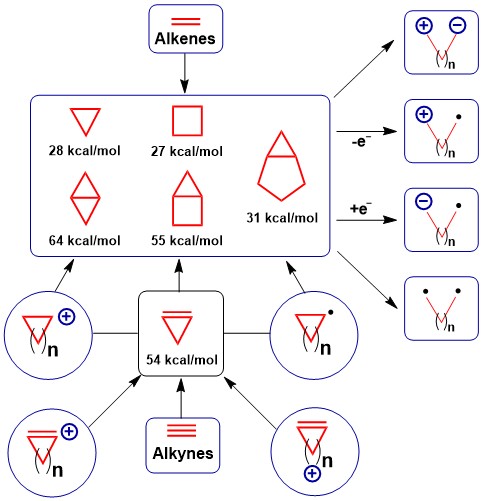
The study of the rigid structure and high strain energy of small rings is one of the oldest and most fascinating fields of organic chemistry (see for example the seminal review of Wiberg: Angew. Chem., Int. Ed. 1986, 25, 312. DOI: 10.1002/anie.198603121). The rigid small rings are finding increasingly application in drug design, and their unique reactivity continue to lead to the discovery of new reactions. Since 2007, our group has been working in small ring chemistry. Initially focusing on the activation of polarized Donor-Acceptor cyclopropanes, we have more recently extended our studies to single-electron pathways and photocatalytic homolytic cleavage for C-C bond activation. We have also developed new methods to access strained rings, with a focus on the use of cyclopropenes as key intermediates. This work resulted in the discovery of new cyclopropenium cation synthons and efficient methods for the synthesis of even more strained bicyclic systems.
Activated cyclopropanes
The ring-strain of cyclopropanes makes them very useful synthetic building blocks in organic chemistry. From the molecular orbitals point of view, they display a high SP2 character (Walsh orbitals) and have therefore often been considered as one-carbon homologues of olefins. The presence of a carbonyl or imine substituent activates cyclopropanes for cyclization and cycloaddition reactions, and this reactivity can be further enhanced by the introduction of a second donor group to give the so-called donor-acceptor substituted cyclopropanes (Synthesis 2009, 3353. DOI: 10.1055/s-0029-1216998). Recently, the activation of other strained rings, such as cyclobutanes, has also begun to be more intensively investigated. In this context, our group is working on the development of new catalytic reactions involving activated small rings, with a particular interest in the use of a nitrogen atom as activating donor substituent (Chem. Commun. 2014, 50, 10912. DOI: 10.1039/C4CC03194F ).
The formal homo-Nazarov reaction

The Nazarov cyclization is the electrocyclic ring-closing of divinyl ketones to form cyclopentenones. Based, on the ring-strain of cyclopropanes, we developed the first catalytic formal homo-Nazarov cyclization (Org. Lett. 2009, 11, 1023. DOI: 10.1021/ol802970g). The catalytic reaction was made possible by the presence of two electron-donating groups (R3 on the cyclopropane and X on the double bond), which stabilized carbocationic intermediates in the reaction. First developed for electron-rich aromatic ring in R3, the reaction was then extended to nitrogen group at this position. At this point, the methodology became useful for the synthesis of aspidosperma alkaloids (Angew. Chem., Int. Ed. 2010, 49, 5767. DOI: 10.1002/anie.201001853).
Regioselective cyclization: Synthesis of goniomitine
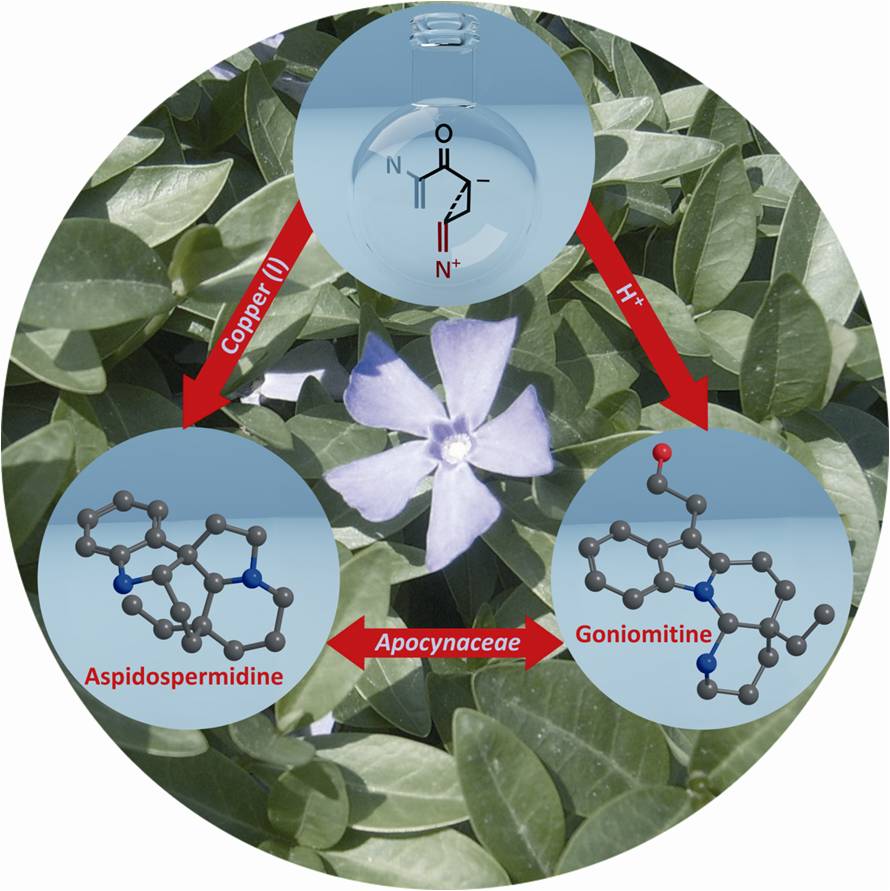
In the case of free indoles as nucleophiles, both cyclization on the C3 carbon or the N1 nitrogen where observed. The regioselectivity of the reaction could be controlled by the choice of solvent and catalysts: Cu catalyst in acetonitrile gave C3, and toluene sulfonic acid in dichloromethane N1 selectivity. This allowed us to accomplish both the formal total synthesis of aspidospermidine and the total synthesis of goniomitine (Angew. Chem., Int. Ed. 2010, 49, 5767. DOI: 10.1002/anie.201001853).
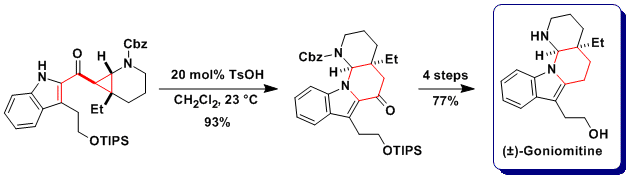
Bioactivity of goniomitine
Despite its very rare structure, there was no report about the bioactivity of goniomitine. In collaboration with Prof. Jürg Gertsch at the University of Bern, we examined the antiproliferative effect of the natural product and several of its analogues. Only a weak cytotoxicity was observed against several cell lines (29-120 microM). Non-cytotoxic alkaloids are relatively rare, and further investigations are currently ongoing to find other biological targets of this rare natural product. This collaboration was initiated in the framework of COST CM0804, an Europe-wide effort towards the study of chemical biology with natural products.
Total synthesis and bioactivity of jerantinine E
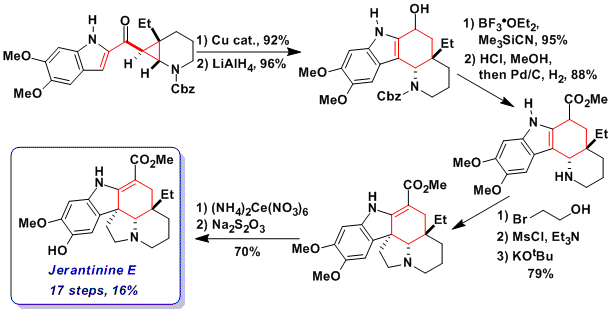
After having achieved the synthesis of goniomitine and aspidospermidine, we wondered if the formal homo-Nazarov strategy could be applied to the synthesis of Jerantinine E, a bioactive alkaloid isolated by Kam and co-workers from the Malayan plant Tabernaemontana corymbosa. This alkaloid had never been synthesized before and its biological mode of action was unknown. The total synthesis of jerantinine E was indeed achieved in 17 steps and 16% overall yield based on four key steps (Angew. Chem., Int. Ed. 2013, 52, 13373. DOI: 10.1002/anie.201305533):
1) Copper-catalyzed formal homo-Nazarov reaction, which proceeded in 92% yield-
2) Installation of the electron-withdrawing ester group via reduction, carbocation generation, cyanation and Pinner methanolysis. The installation of the ester group on an early stage of the synthesis was essential to avoid the decompositions of oxidation sensitive intermediates.
3) Formation of the fifth ring of the natural product via a three-step one-pot bis-alkylation protocol developed by Rawal and co-workers.
4) Selective oxidative deprotection of one of the methoxy group.
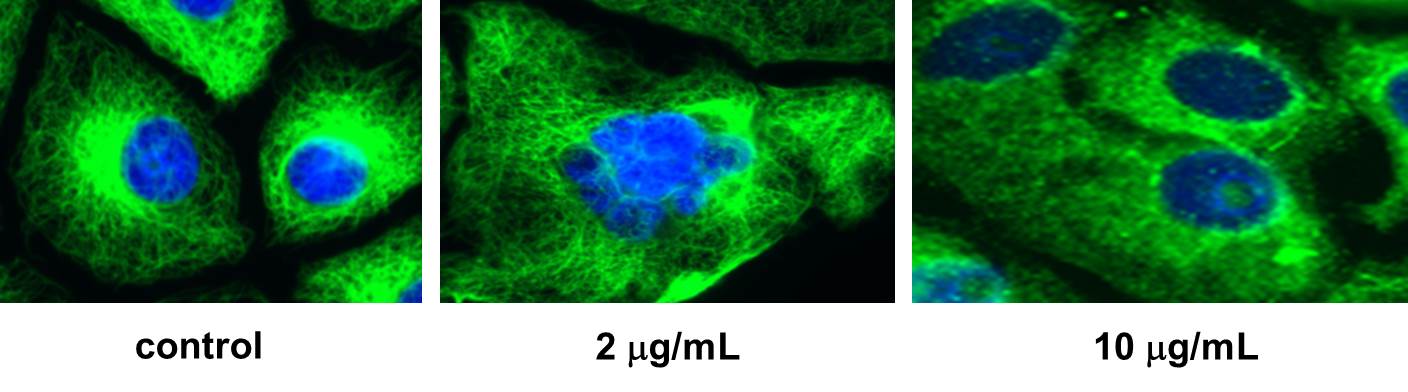
With significant amounts of jerantinine E now available, our collaborators at EPFL (Group of Prof. Sandrine Gerber ) and at the Helmholtz Institute (Group of Dr. Florenz Sasse ) were able to identify tubulin as the target of the natural product. Indeed, an inhibition of the polymerization of tubulin was well visible in presence of jerantinine E.
From cyclization to annulation
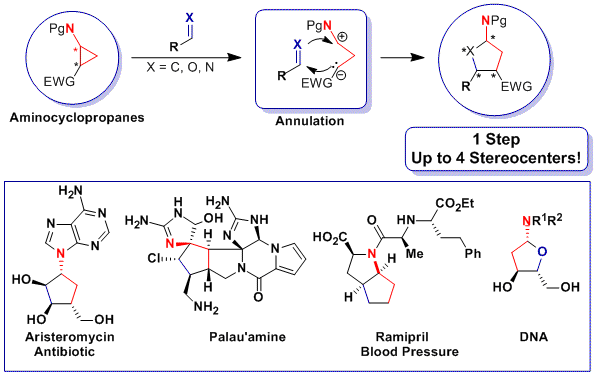
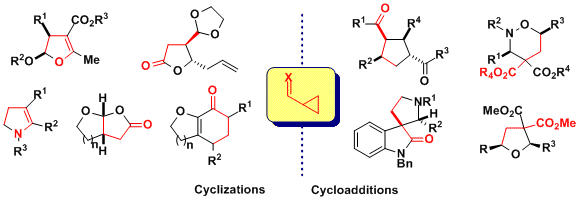
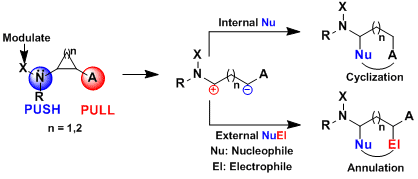
During our work on the total synthesis of natural alkaloids, we discovered the exceptional properties of aminocyclopropanes in cyclization reactions. At this point, we realized that more convergent [3+2] annulation reactions of aminocyclopropanes with olefins and carbonyls were unprecedented. Such processes would have an impressive potential for the synthesis of bioactive natural products, such as aristeromycin or palau’amine, drugs such as ramipril or biomolecules such as DNA and its synthetic analogues. In 2011, we discovered the first catalytic [3+2] annulation reactions of aminocyclopropanes, both with enol ethers (Angew. Chem., Int. Ed. 2011, 50, 12075. DOI: 10.1002/anie.201106255) and aldehydes (Org. Lett. 2012, 14, 386. DOI: 10.1021/ol203144v ) and ketones (Chem. Eur. J. 2012, 18, 4844. DOI:10.1002/chem.201103971 ) . Key for success was the correct modulation of the electronic properties of the donor (phthalimide) and acceptor (diester) groups. In particular, the reactions with enol ethers and ketones were enantiospecific, giving access to enantioenriched cyclopentylamines.
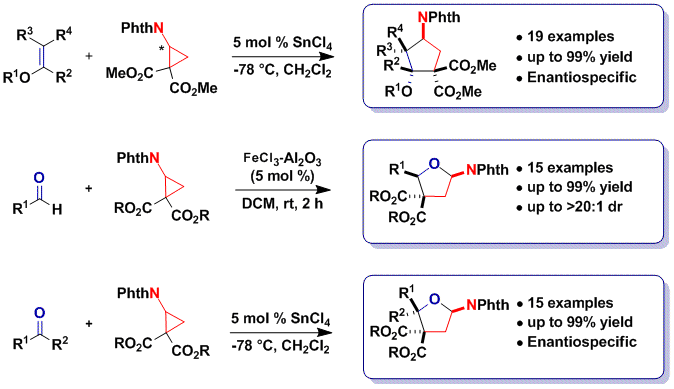
A major limitation of the originally developed protocol was the requirement for succinimide and phthalimide as protecting groups. Cleavage of these groups can be challenging for cyclopentylamines and could not be achieved in the case of tetrahydrofurylamines. On the other hand, nucleoside analogues are a very important classe of bioactive compounds, including already 45 FDA approved drugs. We speculated that thymine-substituted cyclopropanes could display similar reactivity as imido cyclopropanes. Indeed, this was the case, and we were able to develop a [3+2] annulation reaction using thymine, uracil and fluoro-uracil substituted diester cyclopropanes with enol ethers, aldehydes and ketones (Angew. Chem., Int. Ed. 2014, 53, 8484. DOI: 10.1002/anie.201404832). 20 unprecedented nucleoside analogues could be obtained using the method and further functionalized. The compounds have been included at the screening facility of EPFL for the discovery of new bioactivities.
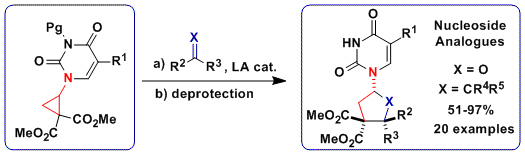
The broad availability of thioalkynes in our group encouraged us to examine this unprecedented class of substrates in [3+2] annulation reactions. Interestingly, two different products were obtained in dependence on the substituent of the alkyne: the expected [3+2] annulation product with silyl groups and an unprecedented polycyclic scaffold with alkyl groups (Chem. Eur. J. 2016, 22, 11997. DOI:10.1002/chem.201602755).
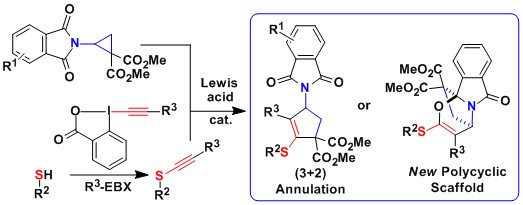
In 2017, the [3+2] annulation of aminocyclopropanes could be extended for the first time to electron-poor N-heterocycles. The reaction worked well for both quinolines and electron-poor pyridines. It was the first time that Donor-Acceptor cyclopropanes could be used to dearomatize efficiently this type of heterocycles. The obtained compounds are important building blocks for the synthesis of indolizidine alkaloids. (Chem. Sci. 2017, 8, 7112. DOI:10.1039/C7SC03197A ).
The exceptional reactivity of aminocyclopropranes is not limited to [3+2] annulation reactions. Simple ring-opening reactions also give access to very useful nitrogen-rich building blocks. For example, the scandium-catalyzed Friedel-Crafts alkylation of indoles and other electron-rich aromatic compounds led to the formation of important gamma-aminobutyric acid (GABA) derivatives (Org. Lett. 2013, 15, 3738. DOI:10.1021/ol401616a ) .
Enantioselective transformations
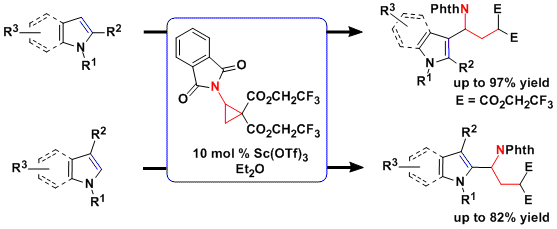
As the [3+2] annulation process is enantiospecific, the synthesis of enantio-enriched products requires the use of enantiopure aminocyclopropanes. However, standard protocols for asymmetric cyclopropanation did not work in the case of vinyl phthalimide. Another possibility would be to develop a dynamic kinetic asymmetric annulation starting from racemic aminocyclopropanes. This approach was successful and enantiomeric ratio up to 98:2 could be achieved using succinimide-substituted cyclopropanes and a cationic copper tBuBOX catalyst with perchlorate as counterion (J. Am. Chem. Soc. 2014, 136, 6239. DOI:10.1021/ja5024578 ). A speculative model for asymmetric induction was proposed based on the distorted square planar structure of copper-tBuBOX complexes: fast attack of the nucleophile is possible only on one of the diastereomeric complexes formed from the racemic cyclopropane; the other complex consequently does not react and ring-opening and racemization can occur.
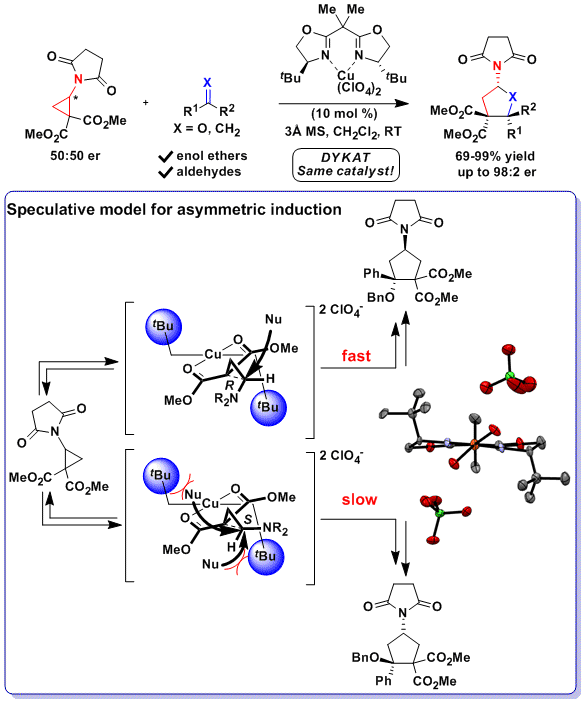
A disadvantage of the DYKAT approach is the challenging control over both reaction kinetics and facial selectivity to achieve high enantioselectivity. In 2018, we reported another approach based on the desymmetrization of meso diaminocyclopropanes. The developed urea-fused cyclopropanes could be used for the highly enantioselective Friedel-Crafts alkylation of indoles. To achieve high enantioselectivity, it was necessary to develop a new bifunctional BOX ligand bearing free hydroxy groups. A this stage, the mode of asymmetric induction is highly speculative. We proposed a model based on hydrogen bonding between the ligand and the diester groups on the cyclopropane, inducing a specific conformation of the tert-butyl groups leading to selective blocking of one of the two reactive positions. (Angew. Chem., Int. Ed. 2018, 57, 5120-5123. DOI:10.1002/anie.201800494)
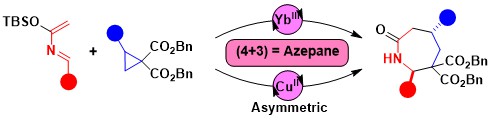
Accessing ring systems larger than six via annulation reactions is much less established than in the case of five- or six-membered rings. One potential approach rely on the use of (4+3) annulation processes. Accessing nitrogen-containing heterocycles is especially interesting, due to their importance in medicinal chemistry. In 2022, we developed a (4+3) annulation of donor-acceptor cyclopropanes and azadienes for the synthesis of azepanones. Both a racemic scandium-catalyzed protocol and an enantioselective method using a copper-Box catalyst could be developed. (Angew. Chem., Int. Ed. 2022, 61, e202209006. DOI:10.1002/anie.202209006)
The field of enantioselective transformations of cyclopropanes is currently in full expansion. In 2020, we published a comprehensive review about this area, describing the key strategies to achieve such processes. (Chem. Rev. 2021, 121, 227-263. DOI:10.1021/acs.chemrev.0c00109)
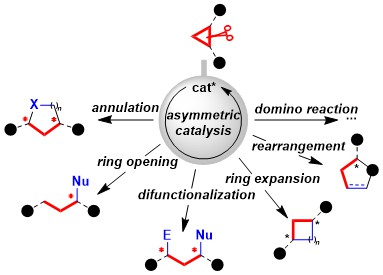
Aminocyclobutanes synthesis and [4+2] annulation
Up to 2013, our work had focused mostly on [3+2] annulation reactions to access cyclopentylamines and tetrahydrofurylamines. We wondered if the polarization strategy successful for cyclopropanes could be extended to cyclobutanes. The resulting [4+2] annulation reactions would give access to cyclohexylamines and tetrahydropyranylamines, which are omnipresent in bioactive compounds. Nevertheless, the required class of donor-acceptor cyclobutanes was unknown. We consequently first developed a new iron-catalyzed [2+2] cycloaddition between enimides and alkylidene malonates (Angew. Chem., Int. Ed. 2013, 52, 9009. DOI: 10.1002/anie.201303803). The method could be used also with substituents on the enimide and on the methylidene malonate to give poly-substituted aminocyclobutanes. Using the obtained aminocyclobutanes, an efficient [4+2] annulation could be developed (Org. Lett. 2015, 17, 1030. DOI:10.1021/acs.orglett.5b00149). The transformation worked for the synthesis of both cyclohexyl- and tetrahydropyranyl- amines. It was also successful for thymine- or uracil-substituted cyclobutanes.
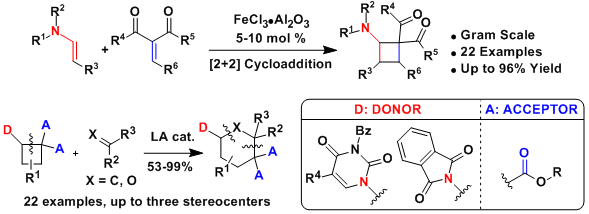
As the [2+2] cycloaddition gave access only to diester-substituted aminocyclobutanes, we were also interested to develop more general synthesis methods. In 2024, we first extended the Ficini reaction to acrylates to access aminocyclobutene esters. Reduction with or without epimerization then lead to trisubstituted aminocyclobutanes with high diastereoselectivity (Chem. Eur. J. 2024, 30, e202401810. 10.1002/chem.202401810).
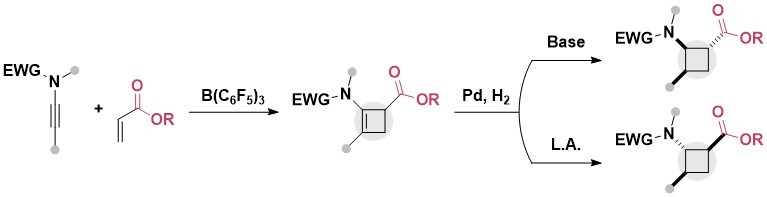
A second method was then developed in 2024 to access N-heterocycle substituted ester cyclobutanes: a Michael addition of N-heterocycles onto in situ generated cyclobutene esters. A main advantage of this method is its simplicity, as simple basic conditions are sufficient to obtain the 1,2-disubstituted cyclobutanes directly from commercially available starting materials (Chem. Eur. J. 2024, 30, e202403986. 10.1002/chem.202403986).
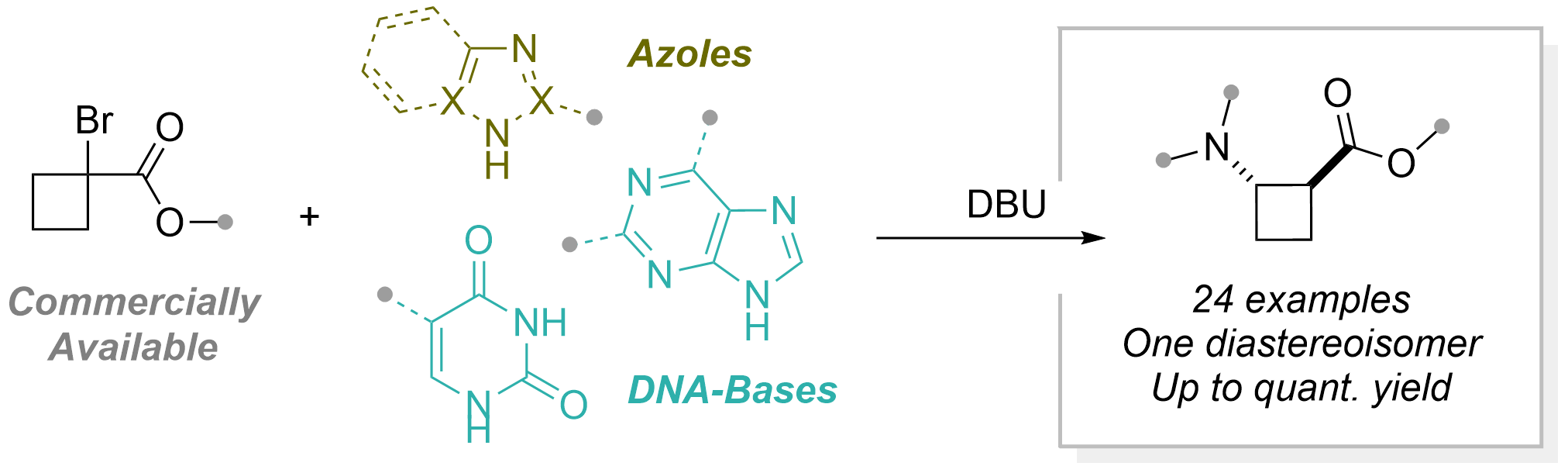
We then turned to thiols as nucleophiles. In this case, isolation of the cyclobutene intermediate was required. We were able to develop a highly enantioselective and diastereoselective method by using chiral bifunctional cinchona-squaramide catalysts (Chem. Sci. 2025, 16, 12115-12121. 10.1039/D5SC01727K).

Mono-ester substituted amino-cyclopropanes and -cylobutanes
When working with DA cyclopropanes, a chelating diester acceptor group is usually required to enable Lewis acid activation. As the diester group is usually not desired in the product, extra steps are usually needed to remove this activating group. In 2021, we reported the efficient activation of mono-ester substituted aminocyclopropanes using silicon Lewis acids. This catalytic system was used in a (3+2) annulation with indoles proceeding with broad scope and high diastereoselectivity (Chem. Sci. 2021, 12, 8706-8712. 10.1039/D1SC01127H).
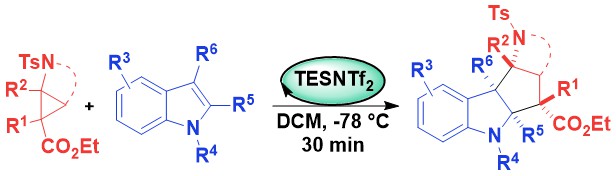
The next logical step was to examine if silylium catalysis would be also applicable to the homologous aminocyclobutanes. However, such substrates had been barely investigated so far and we had to first develop their efficient synthesis based on Michael addition of amines onto in situ generated cyclobutenes. Silylium catalysis was again successful in an annulation reaction with indoles, although it required higher temperature. The intramolecular reaction was especially interesting: At lower temperature, a kinetic annulation product with the core structure of the Malagasy alkaloids was obtained, whereas at higher temperature a different product corresponding to the Akuamma alkaloids was formed (Angew. Chem., Int. Ed. 2023, 62, e202302420. DOI: 10.1002/anie.202302420 ).
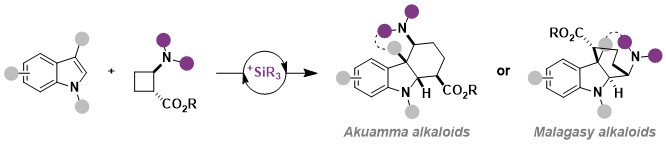
In 2026, we used this methodology for a very short formal synthesis of the natural alkaloid strictamine in only 13 steps (Helv. Chim. Acta 2026, e202500163. DOI: 10.1002/hlca.202500163 ).
In 2026, we also wrote a review on the emerging field of annulation reactions involving Donor-Acceptor cyclobutanes (Helv. Chim. Acta 2025, 108, e202500140. DOI: 10.1002/hlca.202500140 ).
Bicyclobutanes
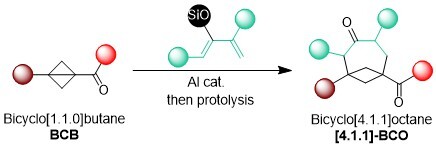
More strained bicyclo[1.1.0]butanes (BCBs) have recently attracted strong in interest in the synthetic community. In 2024, we become also interested in the use of BCBs for accessing larger ring systems. Using silyl enol ethers as partners, we developed a formal [4+2] cycloaddition giving access to rigid bicyclo[4-1-1]octanes in high yield. The chemical space around such medium size rigidified ring systems has been poorly investigated so far and these scaffolds could be therefore of interest for medicinal chemistry. (Chem. Sci. 2024, 15, 10823-10829. (10.1039/D4SC02767A)
Donor-only cyclopropanes
Our research program on Donor-Acceptor aminocyclopropanes has been highly successful. Nevertheless, the presence of the two ester groups can be a drawback, as they are usually not desired in the product and need to be removed. Therefore, we turned to the activation of simple mono-substituted aminocyclopropanes. In 2019, we had a first success in this area. Upon oxidation with N-iodosuccinimide, aminocyclopropanes can be opened to the corresponding highly reactive iodinated imines. The latter constitute an interesting dielectrophilic synthon, in contrast to Donor-Acceptor cyclopropanes, which have zwitterionic reactivity. Nucleophilic substitution and addition then delivered a broad range of functionalized propyl amines. (Angew. Chem. Int. Ed. 2019, 22, 9123-9127. DOI:10.1021/acs.orglett.0c03528 )
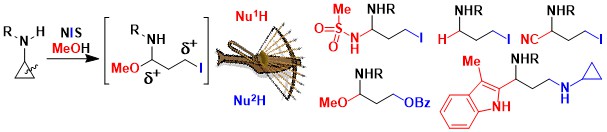
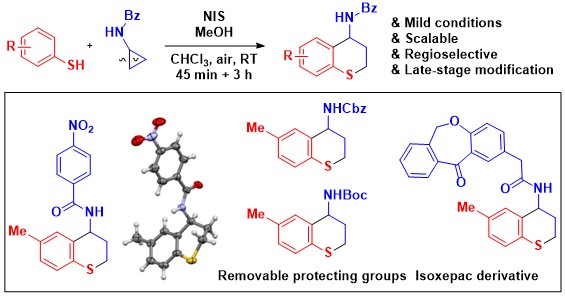
In 2020, we reported that annulation processes were also possible under these conditions: the combination of thiophenols and cyclopropylamides led to the formation of thiochromans in good yields (Org. Lett. 2020, 58, 9123-9127. DOI:10.1021/acs.orglett.0c03528).
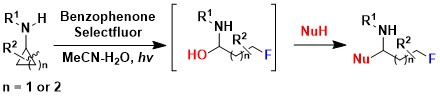
Fluorinated compounds are very important in synthetic and medicinal chemistry. However, we could not introduce fluorine atoms in our first developed method for the oxidative ring opening of aminocyclopropanes. In 2020, we turned to photoredox catalysis to achieve such a transformation: In presence of cheap benzophenone as photocatalyst and Selectfluor as oxidant/fluorine source, an efficient oxidative fluorination of cyclopropylamides was achieved. A range of different nucleophiles could be again added to the formed iminium intermediate (Angew. Chem., Int. Ed. 2020, 59, 16420-16424. DOI:10.1002/anie.201907060).
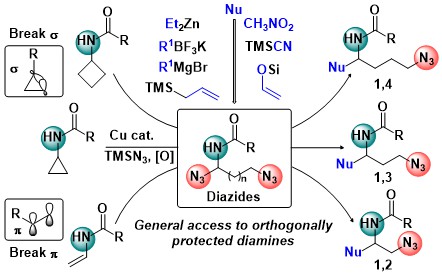
Diamines are another very important class of building blocks for synthetic and medicinal chemistry. In 2021, we developed a copper-catalyzed oxidative diazidation of aminocyclopropanes. In the obtained products, one azide was acting as a masked amine, and the other as a leaving group to generate an imine intermediate, which reacted smoothly with numerous nucleophiles. This method gave a fast access to structurally diverse orthogonally protected 1,3-diamines. The diazidation was also successful in the case of enamides and aminocyclobutanes, leading to the formation of 1,2- and 1,4- diamines respectively (J. Am. Chem. Soc. 2021, 143, 11969-11975. 10.1021/jacs.1c06700).
Our recent work as well as results reported by other research groups have demonstrated the high potential of radical approaches for the functionalization of aminocyclopropanes. This motivated us to review this field in 2022. (Chem. Soc. Rev. 2022, 51, 7344-7357. DOI:10.1039/D2CS00090C).
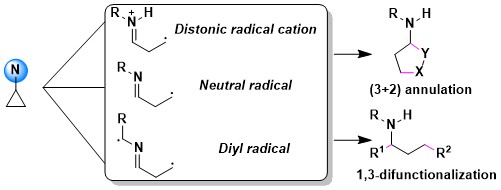
When considering the oxidative properties of hypervalent iodine reagents, using them to functionalize electron-rich cyclopropanes appeared as a next logical step. In 2022, we demonstrated that photoactivated EBX reagents were efficient to promote the oxyalkynylation of both amino- and aryl-cyclopropanes. An interesting unexpected results was a complete switch in reaction outcome for ortho di-substituted aryl cyclopropanes: In this case, an efficient C-H alkynylation of the cyclopropane was observed instead. (Chem. Sci. 2022, 13, 12831-12839. DOI:10.1039/D2SC04344K).
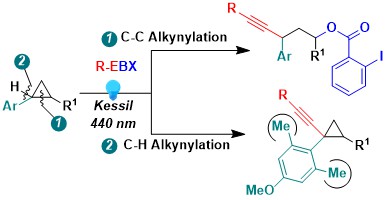
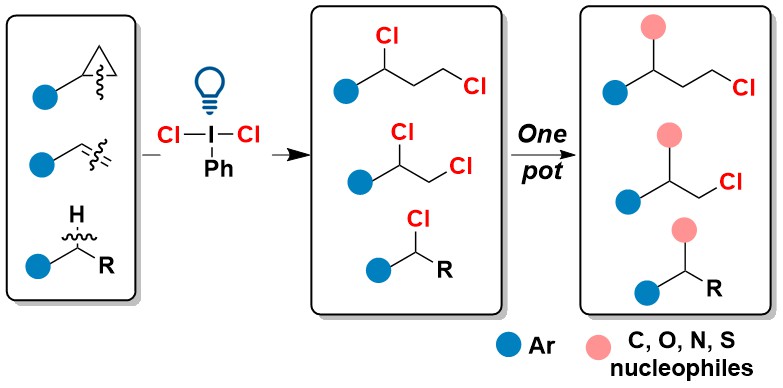
Of course, many other photoactivated hypervalent iodine reagents could be potentially used for the oxidative cleavage of C-C bond in strained rings. In 2026 we reported our results using the oldest of all hypervalent iodine reagents, PhICl2 (Willgerodt’s reagent). This reagent was routinely used for electrophilic chlorination, but we showed that under blue light irradiation, it can become an excellent source of chlorine radicals, enabling not only fast chlorination of C-C bonds in strained rings, but also double bond and C-H chlorination. Preliminary results were also obtained for a method catalytic in iodine using Selectfluor as oxidant, but these conditions were less efficient. (JACS Au 2026, 6, 290-298. DOI:doi.org/10.1021/jacsau.5c01226).
Acceptor-only cyclopropanes
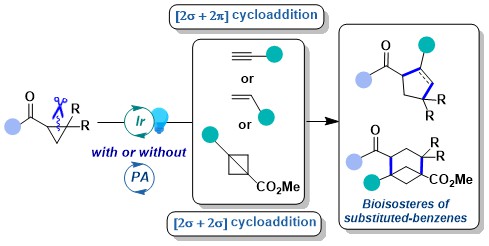
The activation of acceptor-only cyclopropanes under reductive conditions is the logical alternative to the activation of donor-only cyclopropanes via oxidation. In fact, this concept has been intensively investigated by many groups in synthetic chemistry. In 2022, we wondered if an alternative strategy based on energy transfer could be successful for these substrates. Such an approach would not require anymore the use of strong reductive conditions. Nevertheless, the use of energy transfer was reported only for bicyclic systems and substrate racemization prior to our work. In 2023, we demonstrated that energy transfer activation was possible for carbonyl-substituted cyclopropanes for annulation reactions with a broad range of partners including alkynes, alkenes and bicyclobutanes (BCBs). The latter enable a very efficient synthesis of bicyclo[3.1.1]heptanes, an interesting class of benzene bioisosteres. Although we could not elucidate all the aspects of the mechanism, we develop in particular a new trapping experiment with DMPO, supporting the presence of a biradical intermediate (J. Am. Chem. Soc. 2023, 145, 25411-25421. 10.1021/jacs.3c09789).
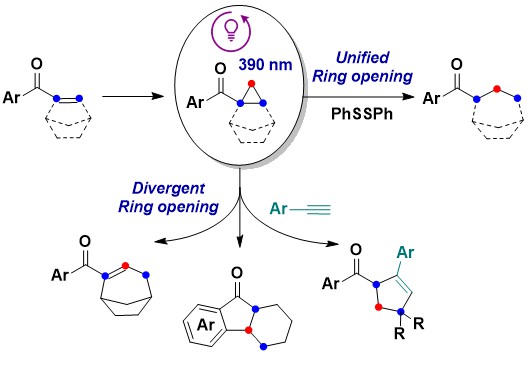
As no electron transfer was required for the activaton of the carbonyl cyclopropanes, direct activation wiht 390 nm UV light became possible even in absence of a photocatalyst. Under these conditions, the outcome of the reaction was substrate dependent, leading in the case of polycyclic cyclopropanes to efficient ring-opening reactions. Based on these results, we were able to develop a general reductive method for C-C cleavage of the more substituted bond of carbonyl cyclopropanes suing PhSSPh as hydrogen source. If combined with a cyclopronation step, this method allowed therefore a one carbon homologation of alkenyl carbonyl compounds proceeding with cleavage of the alpha-beta C-C bond. (Angew. Chem., Int. Ed. 2024, 63, e202411719. 10.1002/anie.202417719).
Cyclopropenes
In 2018, we started a new research program involving a further class of activated small rings: the cyclopropenes. The double bond in these compounds is highly strained and reactive, which appears to us highly promising for developing new transformations. However, several attempts using transition metal or Lewis acid activation were thwarted by concurring polymerization side reactions. In 2019, we were finally successful and reported a new annulation reaction combining aminocyclopropanes and cyclopropenes. Under photoredox conditions, an oxidative ring-opening of the aminocyclopropanes occurred, and the formed radical added rapidly to the double bond of the cyclopropenes. Cyclization then led to bicyclo[3.1.0]hexanes, which are important scaffolds in synthetic and natural bioactive compounds. In the case of difluorocyclopropenes, the reaction proceeded with good diastereoselectivity (Chem. Sci. 2019, 10, 10716-10722. DOI:10.1039/C9SC03790J).
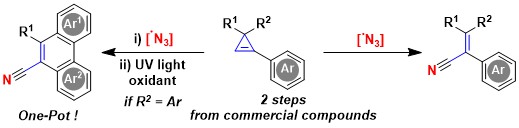
We then wondered if the addition of N-centered radicals to cyclopropenes could also be achieved. Such a process had not been reported before. Based on our experience in the generation of azide radicals, we first decided to examine these species. Indeed, addition of azide radicals to the double bond of cyclopropenes was successful. However, the formed intermediates were unstable and ring opening occurred, resulting in the formation of alkenyl nitriles. An interesting application of such products was an oxidative photocyclization to give polycyclic aromatic compounds, which are important for applications in medicinal chemistry and materials science (Angew. Chem., Int. Ed. 2021, 60, 4075-4079. DOI:10.1002/anie.202013516).
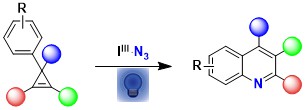
In 2021, we combined two fields of research in our group: azidation with hypervalent iodine reagents and small ring chemistry to develop a new synthesis of quinolines starting from cyclopropenes. In this work, direct activation of ABZ with light in presence of acetoxybenziodoxolone was sufficient to generate the required azido radical (Org. Lett. 2021, 23, 5435-5439. 10.1021/acs.orglett.1c01775).
Up to 2022, our cyclopropene work had focused on the use of radical intermediates. We wondered if potentially even more unstable cyclopropyl cation intermediates could be synthetically useful. There was only very few precedence in the literature that such intermediates could be stabilized in the form of ionium cations with larger atoms, such as iodine or selenium. In 2023, we developed the first semipinacol rearrangement of cyclopropenylcarbinols for the synthesis of highly substituted cyclopropanes. The reaction was successful both for aryl migration and for strained-promoted alkyl migration. The latter case in particular enabled the synthesis of diverse spirocyclic compounds with unprecedented substitution patterns. Iodine, selenium and sulfur-based electrophiles could be used in this transformation (Org. Lett. 2023, 25, 6999-7003. 10.1021/acs.orglett.3c02543).
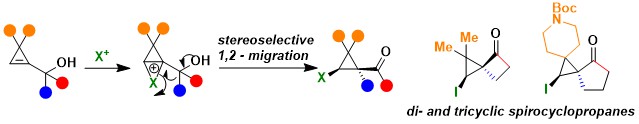
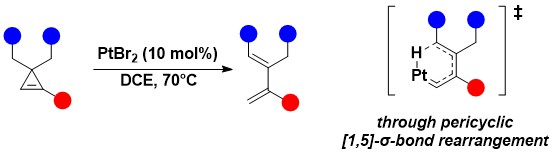
When attempting to use electrophilic platinum complexes to activate the cyclopropenylcarbinols, we serendipitously discovered another transformation: an efficient direct isomerization to dienes. This reaction gave access to highly substituted dienes and was proposed to proceed through a pericyclic [1,5]-sigma bond rearrangement (Helv. Chim. Acta 2024, 107, e2024000105. 10.1002/hlca.202400105).
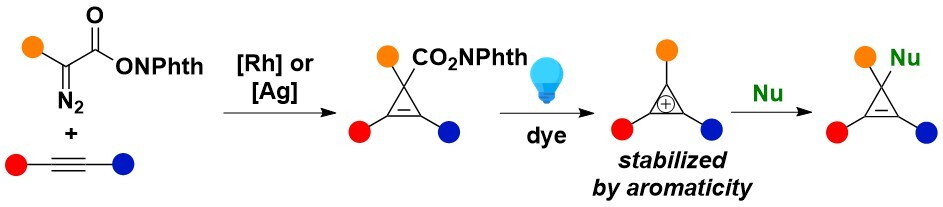
As next potential useful reactive intermediates, we decided to investigate cyclopropenium cations. Since the seminal work of Breslow on these compounds and the discovery of their aromatic stabilization, their practical use had been hampered by the lack of methods to access them with diverse substitution patterns. Our approach was based on a radical-polar crossover reaction from a cyclopropenyl radical generated photochemically. A broad range of nucleophiles could be added to the in situ generated cyclopropeniums, even in the case of non-stabilized mono substituted cations. Indeed, unprecedented substitution patterns could be generated using this approach (Angew. Chem., Int. Ed. 2024, 63, e202404265. 10.1002/anie.202404265). Our work on π-type cyclopropenium cations make us wonder if synthons equivalent to highly unstable σ-type cyclopropenium cations could be also developed. We achieved this goal by introducing CpBX hypervalent iodine reagents, and this work is described in the hypervalent iodine reagents section.
Our cyclopropenium chemistry in particular gave an efficient access to carbonyl compounds bearing a cyclopropene in alpha position by addition of silyl enol ethers. This enabled us to design a new strategy for the synthesis of housanes using a carbometallation-cyclization cascade. We demonstrated for the first time-that extended metallacycles could be used to direct the carbocupration of strained rings. A broad range of alcohol-substituted housanes could be obtained by this method, which are interesting both as cyclopentane isosteres and as reactive strained intermediates for the stereoselective synthesis of cyclopentenes (ChemRxiv 2026. 10.26434/chemrxiv.10001480/v).
