Oxygen Diffusion in Alumina
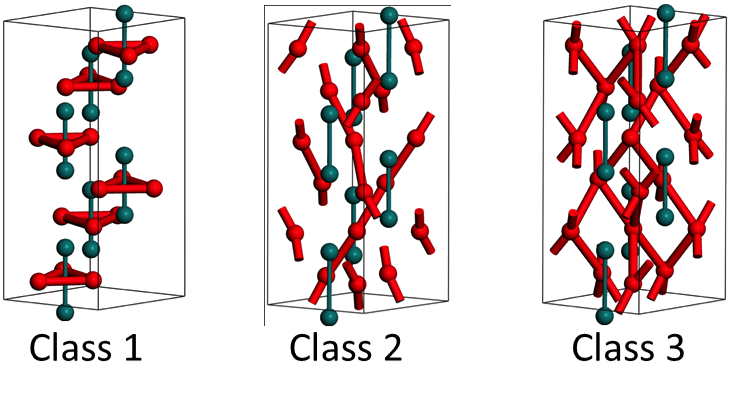
The energy for oxygen vacancy diffusion calculated theoretically (1-2 eV) are significantly smaller than experimentally observed activation energies (5-6 eV)[5]. Atomistic simulations in pure alumina systems have shown different possible diffusion paths with different migration barrier energies (figure below class 1,2,3 jumps) but still not comparable to experimental values.[6] This may be due to impurities, which are always present, even in “pure” materials (i.e. ppm levels of impurities). Consequently the effect of the interaction of oxygen vacancies with Mg impurities was simulated using atomistic defect cluster and nudged elastic band (NEB) methods [6]. We find that oxygen vacancies can form energetically favourable clusters [6], which increases the energy barrier for the detachment of a single vacancy from Mg. This helps explain part of the discrepancy between experimental observations and theoretical calculations. Future modelling work needs to focus on three key issues: (1) taking into account other cation impurities present in experimental alumina, (2) investigating the possibility of tightly bound cluster migration in alumina and (3) developing simulation methods to quantify the effect of impurities on “macroscopic” diffusion in alumina.
 |
|
| Different diffusive jumps for oxygen vacancies in alumina simulated with metadynamics – with migration barriers – 0.52 eV (class 1); 1.07 eV (class 2); and 1.77 eV (class 3)[5,6] |
 |
|
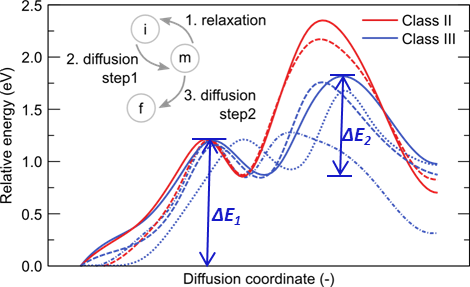
| Typical two step minimum energy pathways for oxygen vacancy jumps as observed in the presence of Mg impurities. The inset shows a schematic view of the relaxation process leading from a metastable (m) position to the new initial (i) position. Going from the new initial (i) position to the final (f) position requires two diffusive jumps via the intermediate metastable (m) state [6] |
Publications
[1] Galmarini S., Aschauer U., Bowen P., Parker S.C., Atomistic Simulation of Y-Doped α-Alumina Interfaces, Journal of the American Ceramic Society, 91(11), (2008), 3643-51, (Link to article)
[2] Galmarini S., Aschauer U., Tewari A., Aman Y., Van Gestel C., Bowen P., Atomistic modeling of dopant segregation in α-alumina ceramics: Coverage dependent energy of segregation and nominal dopant solubility. Journal of the European Ceramic Society (2011),31:2839–52 (Link to article)
[3] Tewari A., Galmarini S., Stuer M., Bowen P., Atomistic modeling of the effect of codoping on the atomistic structure of interfaces in α-alumina. Journal of the European Ceramic Society (2012),32:2935–48 (Link to article)
[4] A. Tewari, F. Nabiei, S. C. Parker, M. Cantoni, M. Stuer, P. Bowen, C. Hébert Towards Knowledge Based Grain Boundary Engineering of Transparent Polycrystalline Alumina Combining Advanced TEM and Atomistic Modeling” J. Am. Ceram. Soc., in press (2015) DOI: 10.1111/jace.13552
[5] Aschauer U., Bowen P., Parker S.C., Oxygen vacancy diffusion in alumina: New atomistic simulation methods applied to an old problem, Acta Materialia, 57(16), (2009), 4765-72, (Link to article)
[6] A. Tewari, U. Aschauer, P. Bowen, “Atomistic Modeling of Effect of Mg on Oxygen Vacancy Diffusion in [alpha]-Alumina” J.Amer.Ceram.Soc., 97(8) 2596-2601 (2014)