From crystal growth and adsorption in cementitious systems to ceramic powders and sintered microstructures.
Responsible : Paul Bowen
Powder Technology deals with powders ranging from a few nanometres (e.g. Superparamagnetic iron oxides for medical applications) through cement and concrete with particle sizes in the 100’s μm range. Control of a materials’ properties comes from being able to master the surfaces and interfaces in the different steps of powder synthesis, crystallisation, dispersion and forming as well as sintering or hardening for cements. Thus the need to understand surfaces and interfaces at an atomistic level rather than treat the materials as a continuum is of prime importance in many areas of research. Experimental methods to access the surfaces and grain boundaries at an atomistic level are often complicated and the resolution limited. This is why simulation at the atomic scale can give important information about structures and surface and interface properties primordial in materials science.
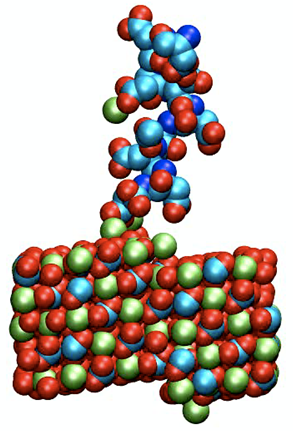
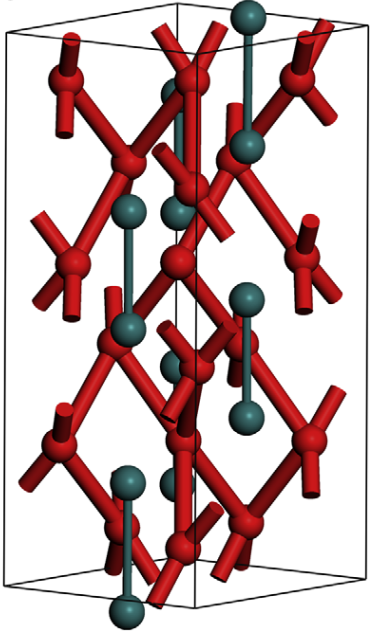
LTP started its effort in atomistic scale simulation in 2005 in collaboration with Prof. Steve Parker at the University of Bath (UK). Our activities on cementitious systems are linked to the industrial-academic research network, nanocem. Within nanocem our efforts are linked with the study of the solid liquid interfaces present in hydrated cement and the development of a force field database for atomistic scale simulations on cementitious materials. For ceramics the structures of grain boundaries in optical ceramic materials for applications such as windows and laser materials has been our main focus. Another field of modelling activity is linked to the adsorption of polymers onto inorganic surfaces that has been a topic of long standing interest for LTP. The interaction of polymeric molecules at the liquid solid interface is of interest both with respect to colloidal stability (a key factor in the dispersion of powders) as well as additives for the control of crystal growth of inorganic powders).
The modelling approach is to use a mixture of energy minimisation, molecular dynamics, metadynamics, nudged elastic band, ab-initio and Monte Carlo (Metropolis as well as kinetic Monte Carlo) simulations to help us model the surfaces and interfaces of materials which will lead to a knowledge based design of microstructures and improved properties.
The Simulation Team are: Dr. Paul Bowen, Dr. Sandra Galmarini and Dr. Abishek Tewari aided and abetted by Dr. Uli Aschauer and our collaboration partners in particular Prof. Steve Parker (University of Bath, UK)
| industrial-academic reasearch network nanocem |