MaD: Macromolecular Descriptors for integrative modeling using cryo-EM data
![]()
MaD (Macromolecular Descriptors) offers an integrative modeling solution focusing on intermediate to low resolution cryo-EM data (5 – 15 Å). Written in Python3, it is as easy to use as selecting components and running it. At lower resolutions, it may be useful to tweak a couple of intuitive parameters.
- Latest version on GitHub: https://github.com/LBM-EPFL/MaD
- (Soon-to-be) submitted version with test data: MaD software and data
MaD is based on local feature descriptors (namely, taking inspiration from common 2D descriptors such as SIFT or SURF). MaD fits structures one at a time using descriptors, which are distinct points in space with an orientation and a vectorial representation. Their efficient matching and clustering enable the transformation of components within the target density with few or no false positives. The associated scoring function is robust to conformational differences between input structures and MaD builds assemblies seamlessly at the end of the procedure, producing in many cases a unique, correct model as output. In case of large structural differences, MaD is able to detect alternate ways of fitting components within the target density, helping further modeling efforts.
Importantly, descriptors are stored in a database for later use (in a running session or later in time). This makes MaD scalable and computationally efficient in general, and more so when including structural ensembles in your integrative project. In this case, a dedicated function highlights the best candidates from the ensemble.
The software is available on the GitHub link above or from the direct link, which also includes testing data in the related article (to be submitted very soon). Testing data includes both simulated and experimental cryo-EM maps at resolutions between 5 and 15 Å.
To generate structural ensembles, we have a preference for molecular dynamics simulations. Their efficient processing for integration within MaD can be done easily using our CLoNe software (see next section on this page).
Clustering with CLoNe
CLoNe is a general clustering algorithm. Here, its use was tailored for structural ensembles, such as those obtained from Molecular Dynamics simulations or integrative modeling attempts. Written in Python3.7, it can be used as any other clustering tool from the Scikit-learn package. When applied to structural ensembles, it will output helpful scripts for automatic loading of the results in the molecular visualization software VMD.
- Latest version on GitHub: https://github.com/LBM-EPFL/CLoNe
- Archive of published version: CLoNe archive
CLoNe is a clustering algorithm with highly general applicability. Based on the Density Peaks algorithm from Rodriguez and Laio (Science, 2014), it improves on it by requiring a single parameter, ‘pdc’, that is intuitive and easy to use. ‘pdc’ can be incremeted if there are too many clusters, and decremented if there are not enough. Integer values between 1 and 10 are usually enough, with many values leading to the same results in most cases.
CLoNe first performs a Nearest Neighbour step to derive the local densities of every data point. Putative cluster centers are then identified as local density maxima. Then, CLoNe takes advantage of the Bhattacaryaa coefficient to merge clusters if needed and relies on a Bayes classifier to effectively remove outliers.
CLoNe was published in Bioinformatics in August 2020:
Lipid Builder
LipidBuilder is a webserver which allows to create lipid bilayers models with atomic resolution, suitable to be used as input for molecular dynamics simulations
LipidBuilder generates automatically the topology and template of a given phospholipid. First, the lipid’s topology is created by combining the selected phospholipid head groups, extracted from a built-in library of structures and the provided hydrocarbon chains. Since the hydrocarbon chains have been parameterized based on a “plug and play” philosophy in the CHARMM force field, the acyl chains are generated by linking a series of alkanes. Three different classes of hydrocarbons have been defined: the saturated, the unsaturated and the cyclopropane. Finally, the psfgen algorithm and the generated topology file are combined to build the template PDB and PSF files of the phospholipid.
Development:
- T. Lemmin*, C. Bovigny*, D. Lancon and M. Dal Peraro, Cardiolipin Models for Molecular Simulations of Bacterial and Mitochondrial Membranes, Journal of Chemical Theory and Computation, 9(1):670, 2013
power: a parallel optimization workbench to enhance resolution in biological systems
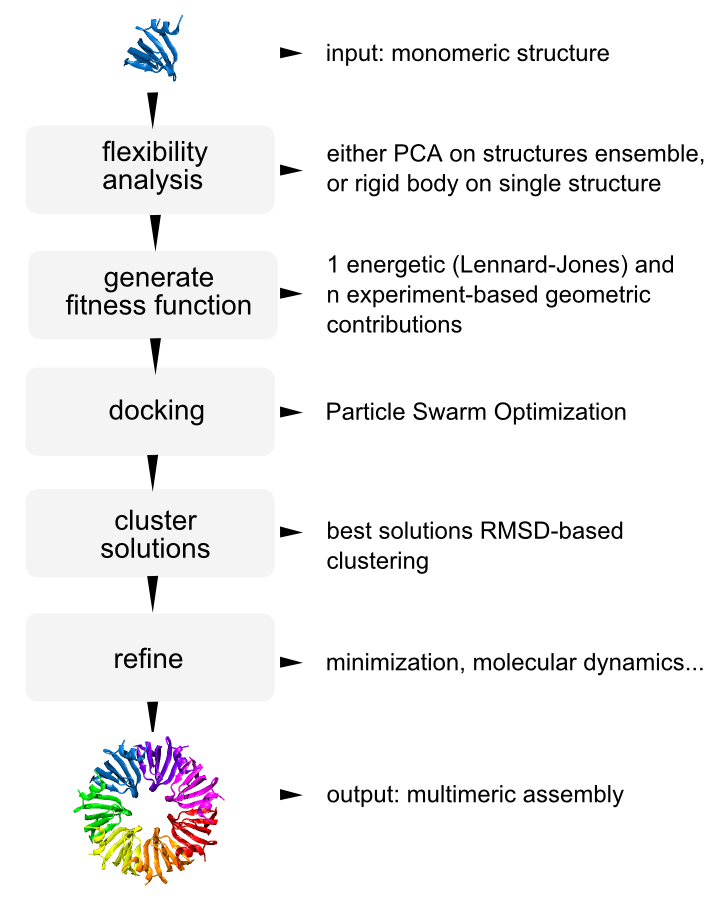 power is an open source optimization framework designed for dynamic integrative modeling of biological systems. In its current version is based on particle swarm optimization and can approach the prediction of molecular assemblies based on a limited set of experimental spatial restraints.
power is an open source optimization framework designed for dynamic integrative modeling of biological systems. In its current version is based on particle swarm optimization and can approach the prediction of molecular assemblies based on a limited set of experimental spatial restraints.
power has been concieved to be modular, thus that the creation of specific modules is easy even for a user unaware of its internal architecture.
The current version supports function minimization, experiment-driven prediction of protein hetero-multimers (see pipeline on the right), and parameterization of multiscale models for molecular simulations.
Development:
- power : a parallel optmization workbench to enhance resolution in biological systems, in preparation.
- M.T. Degiacomi and M. Dal Peraro, Macromolecular Symmetric Assembly Prediction Using Swarm Intelligence Dynamic Modeling, Structure, 2013, 21(7), 1097.
Applications:
- Degiacomi MT, Iacovache I, Pernot L, Chami M, Kudryashev M, Stahlberg H, van der Goot FG, Dal Peraro M, Molecular assembly of the aerolysin pore reveals a swirling membrane-insertion mechanism. Nature Chemical Biology 9, 623–629 (2013).
- Kudryashev M, Stenta M, Schmelz S, Amstutz M, Wised U, Castaño-Díez D, Degiacomi MT, Mannish S, Beck CKE, Kowal J, Diepold A, Heinz DW, Dal Peraro M, Cornelis GR, Sahlberg H, In situ structural analysis of the Yersinia enterocolitica injectisome, eLife, 2:e00792. doi: 10.7554/eLife.00792 (2013).
last update: 04.07.2013
Crowding
Interactive plot with CSP data from Biophys J 2016 is available at this link:
http://lucianoabriata.altervista.org/papersdata/crowding2016.html