Molecular simulations are unique tools to observe processes at otherwise unreachably high combined spatio-temporal resolution. In addition, they can simultaneously be used to estimate macroscopic thermodynamic data, such as diffusion coefficients, mass transfer coefficients, activity or surface tension, which are related to cloud droplet formation and can often be directly imported as an input cloud droplet nucleation models. Our focus is to provide methods to estimate such thermodynamic data in liquid, semisolid and glassy states, use them in macroscopic models and link resulting effects on cloud microphysics back to molecular structure, using equilibrium MD as well as state of the art enhanced sampling techniques such as Metadynamics or Steered MD.
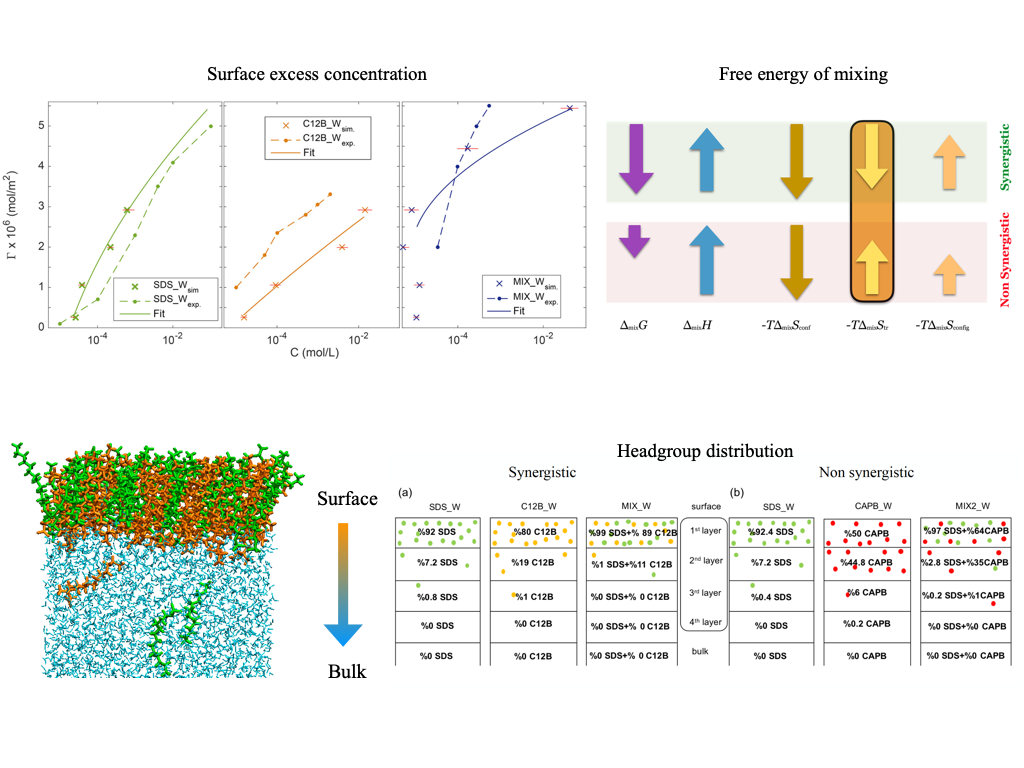
Surface tension – synergism and molecular structure in ionic surfactant mixtures
Surface tension of atmospheric aerosol may reach very low values, which can have drastic effects on cloud droplet activation. According to one hypothesis, low surface tension is due to the presence of bioaerosol particles, while another claims that it is caused by the synergistic enhancement of surface tension reduction in mixed surfactant systems. We use thermodynamic integration MD to assess the plausibility of the latter by comparing the surface excess free energies for pure and mixed synergistic and non-synergistic surfactant systems. Estimation of entropic and enthalpic contributions to the free energy of mixing reveals that the sign of the two-body excess entropy determines whether a pair of surfactants form a synergistic mixture or not. With the help of intrinsic surface analysis, we find that, in terms of structure, synergism is mainly due to the close packing of surfactant headgroups and the increased order of the carbon chain. Any branching observed in the alkyl chain of the surfactant reduces close packing, and thus the synergistic contribution to surface tension reduction. Since under atmospheric conditions a mixture of numerous different surfactants are present on the droplet surface — among them very likely highly branched ones (eg.:HULIS), we conclude that synergistic behaviour of surfactant mixtures is unlikely the main cause of reduced surface tensions in atmospheric aerosol.
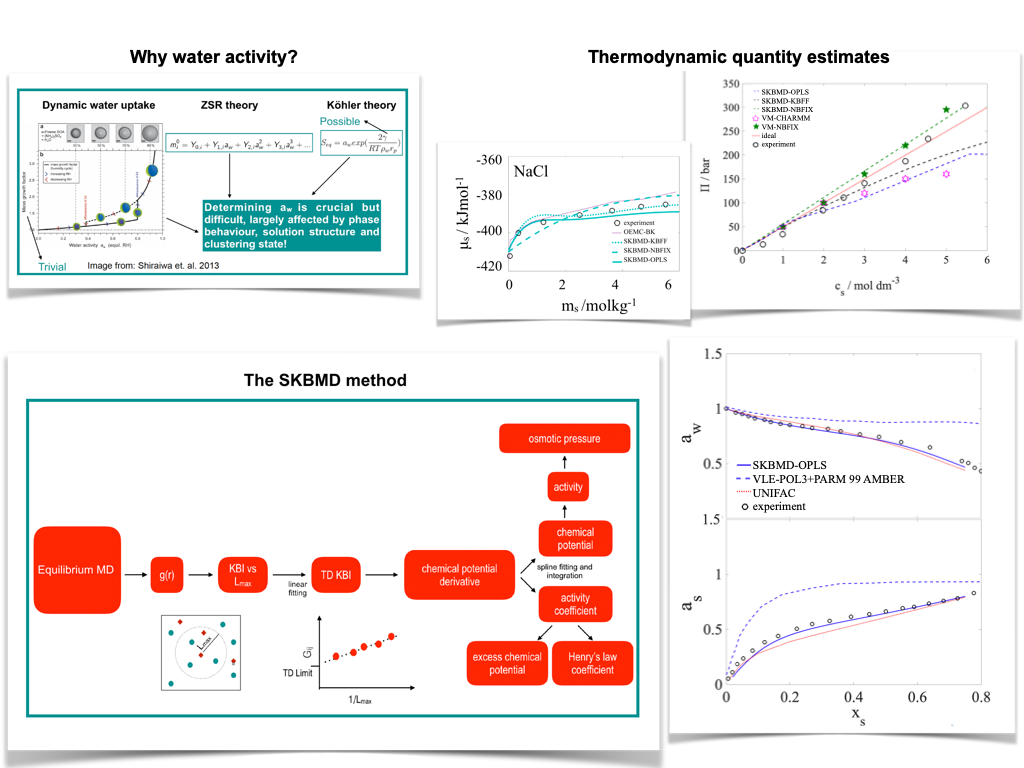
SKBMD – a simple method to estimate activity from equilibrium MD simulations
Water activity plays a central role in determining multiphase partitioning via the Kohler and Gibbs adsorption theory and the linear isopiestic relations of Zdanovskii, Stokes and Robinson. We have developed a simple method to estimate water activity and related quantities, such as osmotic pressure or chemical potential, via MD simulations using the Kirkwood-Buff theory in the subvolume approximation (SKBMD). The SKBMD method provides remarkably good estimates (within 2-5% percent of the experimental results) of the water activity for aqueous solutions from close to ideal (NaCl/water) to strongly non-ideal (ethanol/water) along the whole miscibility range. Despite its simplicity, the performance of SKBMD in reproducing activity is comparable to other simulation techniques of higher computational complexity, and is significantly higher when compared to other methods based on simple equilibrium simulations.
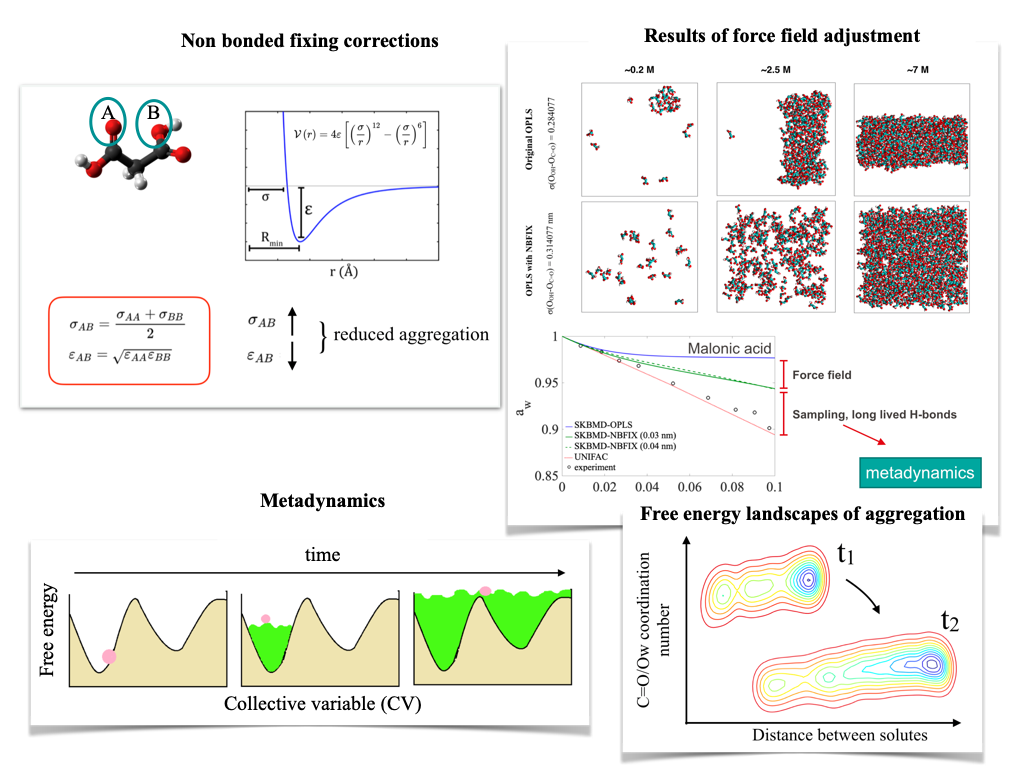
SKBMetaD and improved force field parametrisation – better sampling for aggregating organics
Dicarboxylic acids represent a large fraction of oxidation end products of organic material in the atmosphere. Due to their relatively low volatility, these compounds tend to partition into the particulate phase, where they exert a strong effect on droplet growth and CCN activity., thus understanding their behaviour in concentrated aqueous solutions is a crucial question. While MD simulations with portable force fields are widely used to study dicarboxylic species, we point out that this approach is prone to give biased estimates of macroscopic thermodynamic properties. Water activity estimates using the SKBMD method reveal that the formation of large and long-lived aggregates may corrupt reliable estimation of thermodynamic properties. This is partially due to portable force fields overestimating the strength of hydrogen bonds between carboxyl groups. In addition, the lifetime of the aggregates inhibits proper sampling of association-dissociation processes within timescales achievable with standard MD techniques. To remedy the first problem, we introduce non-bonded fixing (NBFIX) corrections — surgical changes in the pairwise non-bonded interaction potential — which effectively reduces aggregate sizes. In order to resolve the second problem, that of lifetimes, we propose a method based on Metadynamics (SKBMetaD), which uses SKB estimates of the water activity to assess whether solution structure is close enough to reality, and thus ensures the credibility of thermodynamic property estimates.
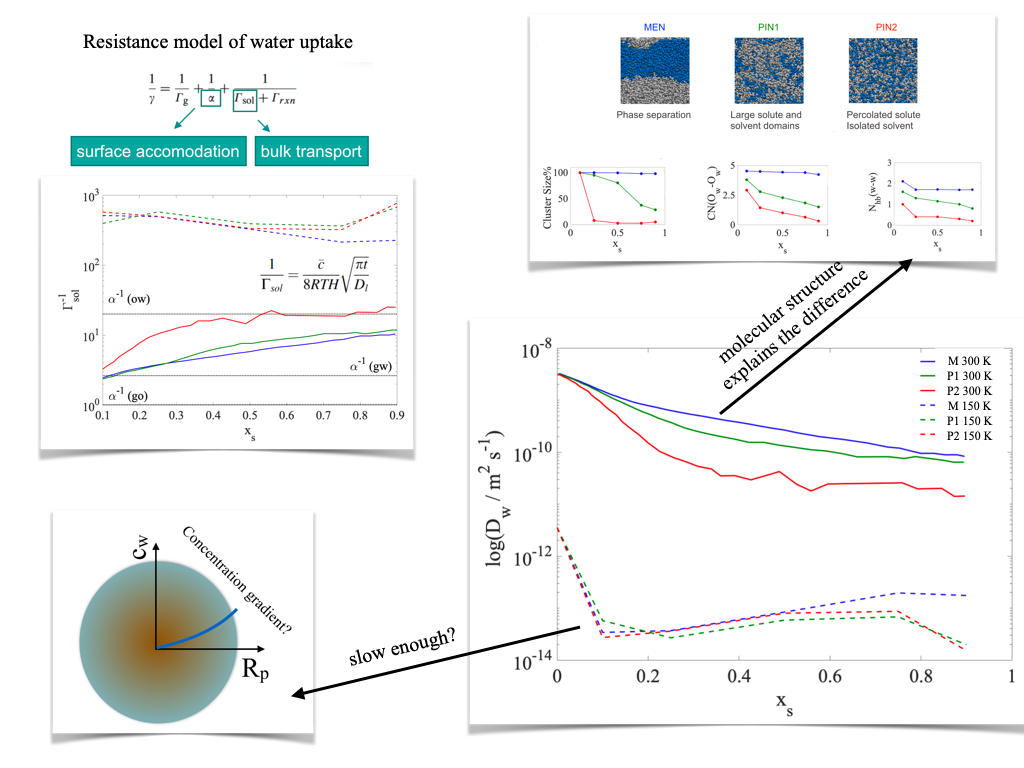
Diffusion of water in liquid and semisolid binary mixtures
The rate of gas-to-particle partitioning, which is closely related — via its effect on particle size — to CCN activity, is determined by surface accommodation, gas phase diffusion and diffusivity in the particle phase. Slow diffusion in in the particle phase can inhibit the growth of semisolid and glassy particles, and can result in radial concentration gradients, which affects vapour pressure, and thus critical supersaturations. Despite of its importance in interpreting experiments and thermodynamic model calculations, the exact role of particle phase diffusivity in aerosol growth mechanisms is to date not fully understood. We use equilibrium MD simulations to estimate diffusion coefficients of water and solutes in binary mixtures of water and α-pinene SOA products at varying concentrations, at 300 and 250 K to model laboratory and tropospheric conditions. Diffusion coefficients of water at room temperature suggest that the systems are in the liquid phase and show a strong concentration dependence that is determined by molecular structure of the solute. At low temperature, diffusion coefficients are approximately 3-4 orders of magnitude lower than at room temperature, indicating a semisolid behaviour; and lack strong dependence on solute structure. Kinetic and hydrogen bonding analysis reveals that at standard temperature the motion of water molecules is more effectively slowed down by solutes which are able to break water’s hydrogen bonded network, while at low temperature this effect is overcompensated slow solute kinetics in general. Diffusion timescales of water range between milliseconds and seconds at standard temperature, while they increase to hundreds of seconds at low temperature.
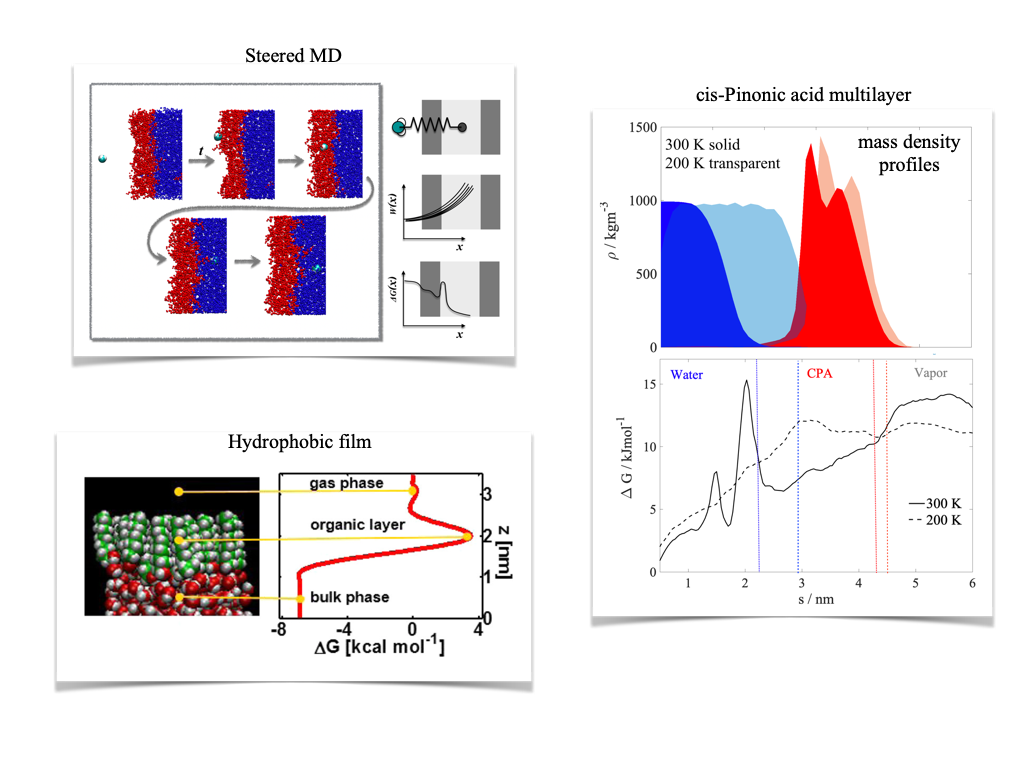
Mass transfer through the gas/particle interface
Accommodation of molecules on the aerosol surface has a strong influence on overall mass transfer kinetics between the gas and the particle phase. Mass accommodation coefficients of water can be very reduced if a continuous hydrophobic organic film covers the surface. Less trivial is the effect of non-ideal multilayers of organics formed when aqueous aerosol undergoes liquid-liquid phase separation. While slow surface accommodation is not considered as a limiting factor for water uptake, recent modelling and experimental studies point out the need for a detailed description of mass transfer kinetics in order to achieve better closure between measured and modelled CCN activities in complex aerosol particles composed of secondary organic aerosol mixed with soluble inorganic salts. We studied the effect of both hydrophobic organic films and more hydrophilic organic multilayers on the surface accommodation coefficient of water using umbrella sampling and steered MD simulations. In hydrophobic films carbon density, which is inversely proportional to the level branching in the carbon chain, determines mass accommodation kinetics. A completely straight chained film of 1-decanol results in an accommodation coefficient of 10-3, low enough to have a strong effect on CCN number concentrations. Less hydrophobic, liquid multilayers of cis-pinonic acid do not reduce surface accommodation coefficients of water but a free energy barrier at the interface of the organic and the aqueous phase results in a mass accommodation coefficient of 0.05, which highlights the importance of considering bulk accommodation when describing mass transfer kinetics.