A. Ortega-Guerrero, M. Fumanal, G. Capano, I. Tavernelli, and B. Smit, Insights into the electronic properties and charge transfer mechanism of a porphyrin ruthenium-based Metal-Organic Framework Chem Mater (2020) DOI: 10.1021/acs.chemmater.0c00356
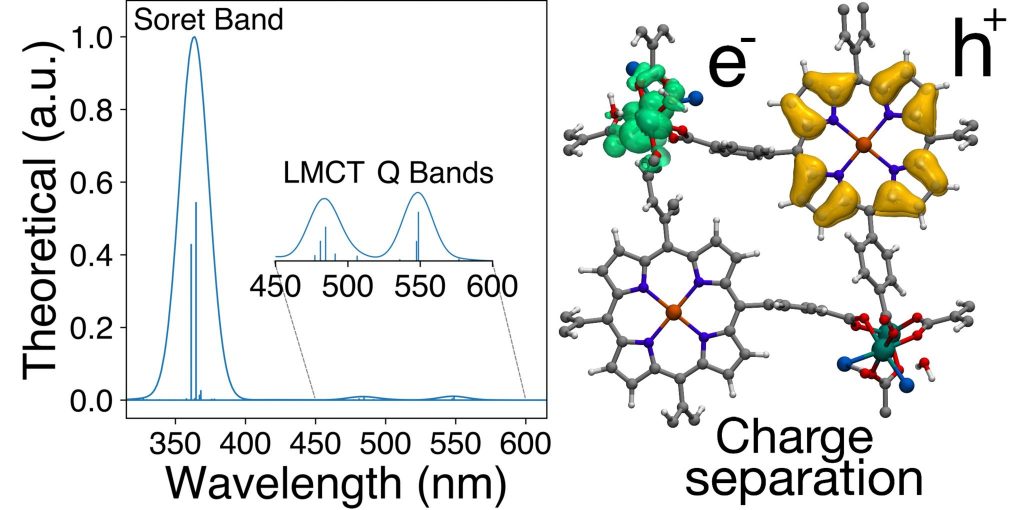
Abstract: Metal-organic frameworks (MOFs) have attracted significant attention in the field of solar-driven photo-catalysis. Recently, a porphyrin ruthenium-based MOF (Ru-TBP-Zn) has shown highly efficient co-catalyst-free photocatalytic hydrogen evolution reaction(HER) under visible light in neutral water. However, a detailed molecular understanding of the electronic and optical properties is missing. In this work, we have conducted Density Functional Theory (DFT) simulations to study these properties in Ru-TBP-Zn. Our DFT calculations indicate instability in the experimentally reported structure of Ru-TBP-Zn. Such instability is resolved by proposing two structural models where Cl or OH-anions are coordinated with the metal backbone of Ru-TBP-Zn. Based on these models, the electronic and optical properties in Ru-TBP-Zn are analyzed. UV/Vis spectra calculations allow identifying the importance of the charge-transfer bands. According to our simulations, two possible charge transfer mechanism can co-exist: the direct photo-induced electron transfer from the porphyrin to the ruthenium upon light absorption, and the relaxation from the optically excited porphyrin to the low-lying ligand-to-metal charge-separated state. The interaction energy of the photo-generated electron-hole carriers is computed considering hole-phonon-electron contributions according to the polaron model. Our calculations predict a repulsive electron-hole interacting energy indicating a low electron-hole recombination rate, which is a prerequisite for multi-electron transfer processes such as HER. The understanding of the electronic properties and charge transfer mechanism in Ru-TBP-Zn paves the way for designing efficient porphyrin-based MOFs for photocatalysis.