Computational protein design mimics natural evolution in silico and provides a powerful approach for creating proteins with novel functions not explored by nature. We typically cycle between calculations in our dry lab and experiments in our wet using in vitro biochemical, biophysical and cellular assays lab for validating our predictions. We also complement in silico design calculations with experimental directed evolution approaches. Lastly, we also collaborate with external laboratories expert in structural biology (S. Prosser, O. Ernst, L. Kay at U Toronto), cellular signaling (M. Bouvier at U Montreal) and protein evolution (E. Procko at U Illinois Urbana Champaign).
1. Computational design of protein interactions and networks
Selective protein-protein recognition is central to a large number of cellular functions. Creating novel functions often requires the reprogramming of protein-protein interactions.
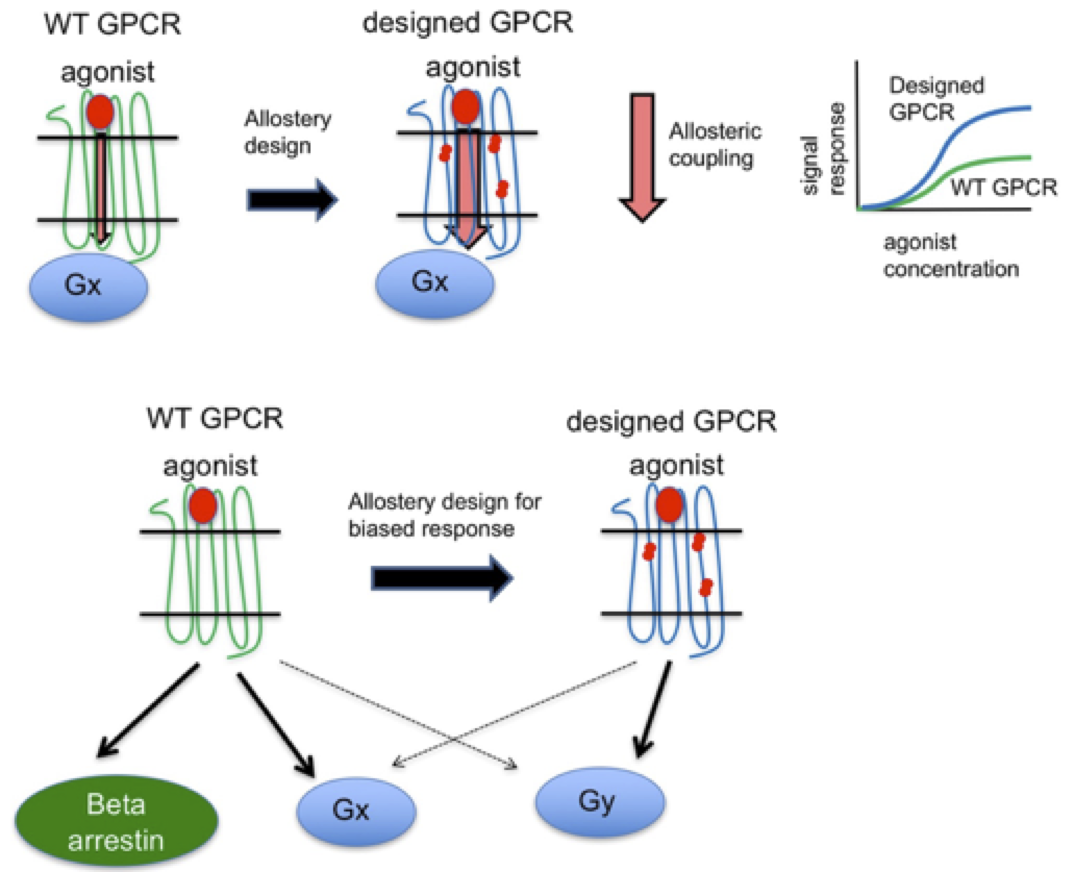
We have developed methods to accurately model protein electrostatic properties (Barth et al., PNAS 2007b) and design highly specific protein-protein interactions (Barth et al., JACS 2008). Using these methods, we designed highly selective peptide inhibitors of important cellular interactions including the essential yeast septins (Barth et al., JACS 2008). We recently implemented a similar approach within RosettaMembrane to design highly specific G protein coupled receptor-G protein signaling pairs (Young et al., under review). These methods pave the road for engineering protein interaction modules or networks with novel signaling specificities for innovative cell engineering approaches.
2. Computational design of membrane protein structures and functions.
Membrane receptors transduce signals across lipid membranes and trigger selective intracellular signaling pathways. As such, they provide key gateways and hubs for the control of cellular functions. Reprogramming membrane receptor functions would considerably leverage synthetic biology and therapeutic approaches that aim at harnessing cells with expanded capabilities.
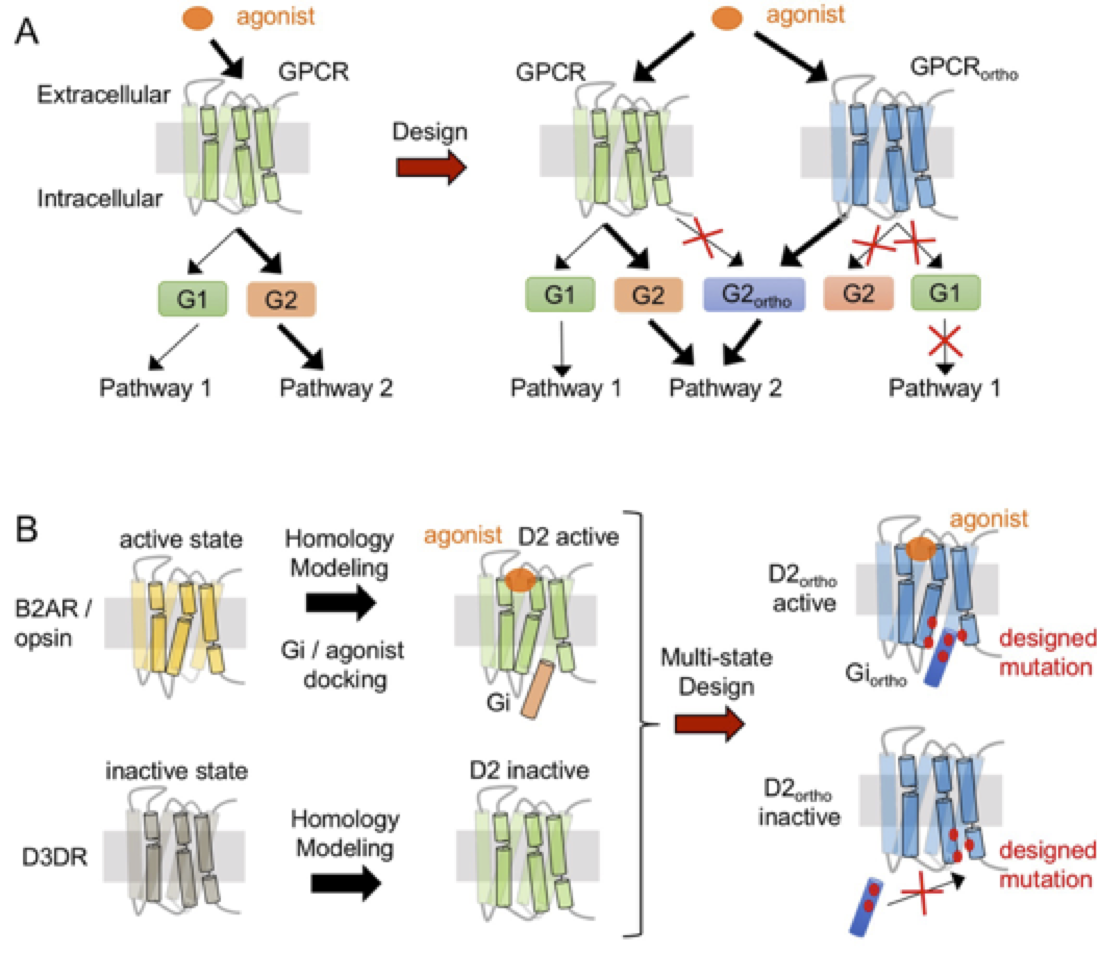
In recent years, we have developed RosettaMembrane for designing membrane proteins. Using the technique, we, for the first time, successfully thermostabilized a G protein coupled receptor and rationally reprogrammed its pharmacological properties (Chen et al., PNAS 2012). During this study, we uncovered common and selective determinants of GPCR metastability shedding new light on how receptor sequences control structural flexibility and signaling properties (Senes & Barth, Nat Struct Mol Biol 2016). We implemented an allosteric model of receptor signaling in RosettaMembrane using which we engineered dopamine D2 receptor variants with rationally altered signaling properties (Chen et al., under review; Keri et al., in prep). We developed an integrated homology modeling-ligand docking-protein design approach that enabled the design of functional dopamine D2 receptors binding and responding to ligands with novel selectivity (Feng et al., Nat Chem Biol 2017). Lastly, we modulate the oligorimerization propensity of membrane receptors by design to uncover the role of receptor associations and create receptor complexes with biased functions (Feng et al., in prep).
Our integrated computational/experimental approaches set the stage for stringently testing our understanding of the allosteric determinants of GPCR signaling, for designing receptor variants thermostabilized for structural studies, or with altered ligand binding and signaling responses for innovative cell signaling studies.