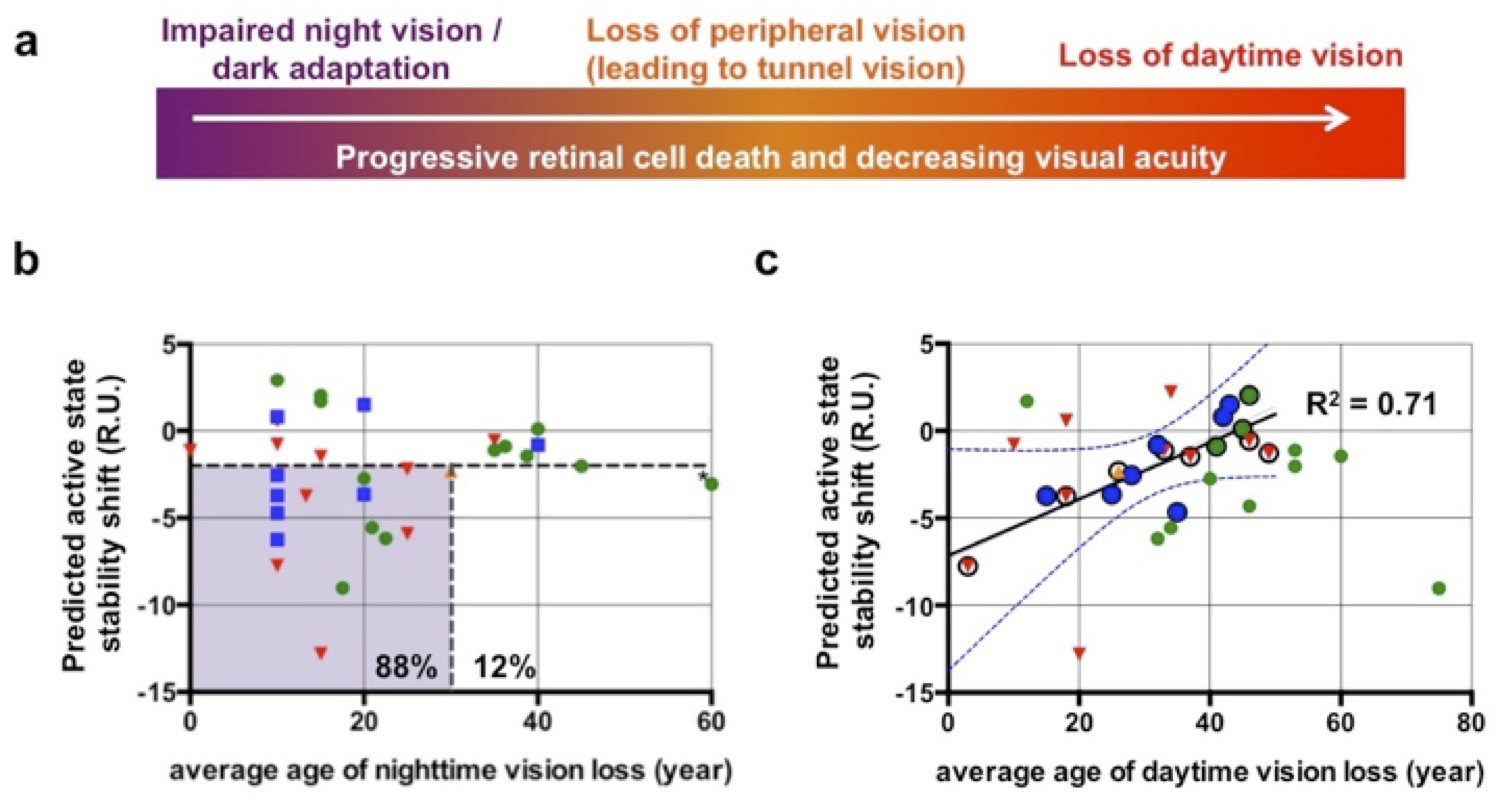
Membrane proteins modeling
1. Modeling membrane protein structures and their interactions with small drug, lipid and peptide molecules.
Membrane proteins constitute around 30 % of all genome encoded proteins, perform critical function in living cells and represent close to 60% of drug targets, but they are difficult to study experimentally and their structures remain poorly characterized. Many membrane proteins undergo conformational changes when they transport substrates or transmit signals across membranes upon sensing extracellular molecules. Conformational flexibility is a key factor encoding the specificity of substrate transport and signal transduction. However, characterizing membrane protein movements remains a challenge for both experimental and modeling approaches. To address these limitations, we strive to develop computational techniques for predicting membrane protein structures and how they respond to the interactions with small drug, lipid and peptide molecule regulators of protein function.
In the past decade, we have developed an ensemble of methods (based on RosettaMembrane) within the software package Rosetta to predict membrane protein structures with unprecedented accuracy:
1) We developed an efficient physical model to recapitulate accurately sequence, structure and energetic properties of transmembrane proteins (Barth et al. PNAS 2007a);
2) Using this model, we predicted near-native structures of large membrane proteins with complex topologies from sequence information alone (Barth et al., PNAS 2009);
3) By combining these techniques with sparse experimental data, we modeled at atomic-resolution specific functional states of the voltage-gated potassium channel and of the full-length a2bIII integrin receptor (Pathak et al., Neuron 2007; Barth et al., Mol Cell 2009). These models reconciled apparently contradicting experimental observations and shed unprecedented light into the molecular mechanisms underlying the function of these receptors. More recently,
4) We further developed RosettaMembrane to predict membrane protein structures by homology to existing structures with high accuracy (Kufareva et al., Structure 2011; Chen et al., Plos Comp Biol 2014). 5) We integrated homology modeling and ligand docking to accurately model ligand-bound GPCR structures (Kufareva et al., Structure 2014; Feng et al., Nat Chem Biol 2017). 6) We implemented a hybrid solvation model in RosettaMembrane to explicitly model and design solvent-mediated interaction networks in membrane protein structures which critically control conformational changes and allosteric functions (Lai et al., Structure in press). Importantly, these modeling techniques expand the number of membrane proteins that can be engineered using rational design approaches and that can be targeted using rational drug design strategies.
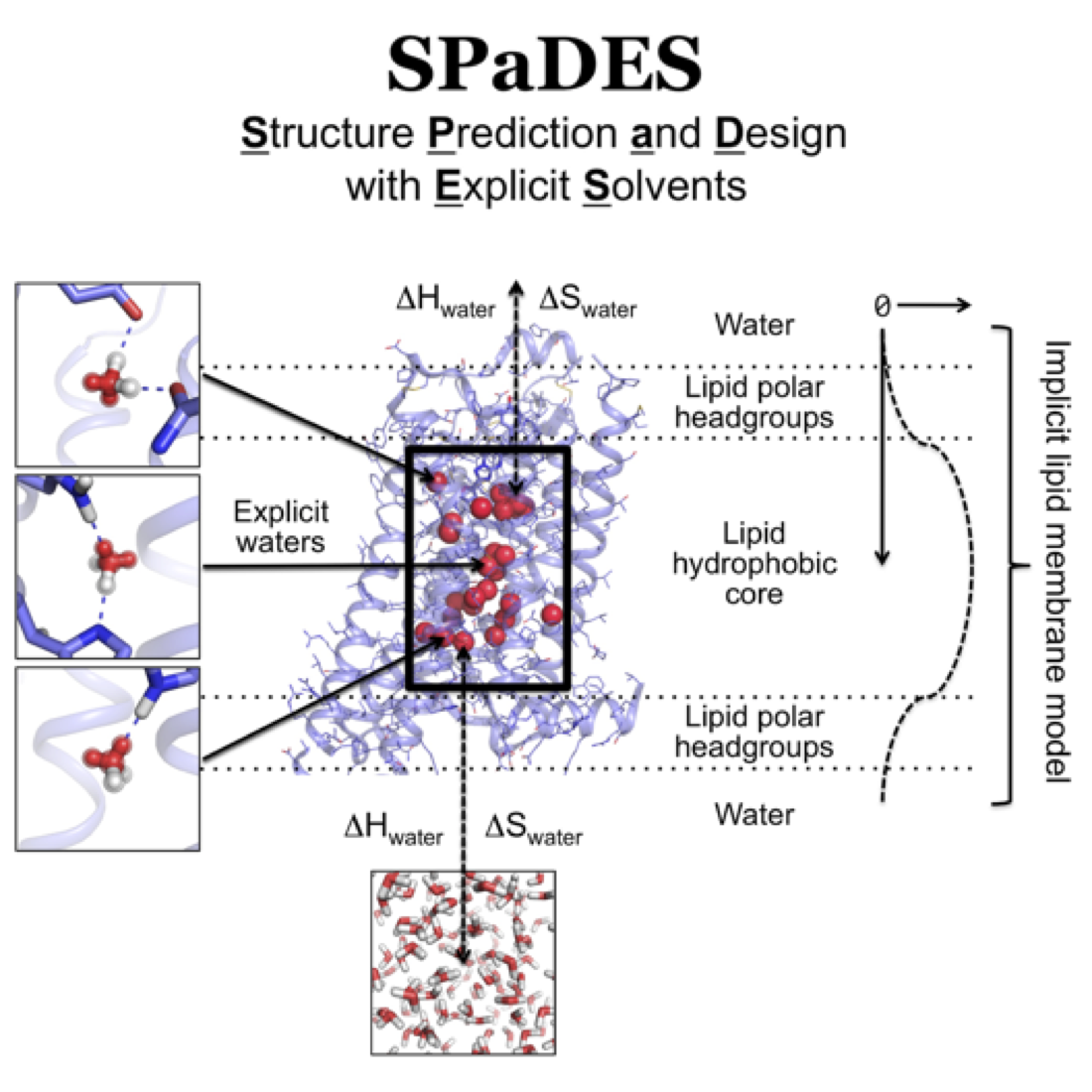
Membrane proteins structures and functions
2. Uncovering principles of membrane protein structures and functions through analysis of genome sequences and protein structure databases.
Growing databases of genome sequences and protein structures provide invaluable information from which sequence/structure relationships coding protein topology, stability and evolution can be extracted. We strive to uncover universal structure/function principles by identifying consensus residue interaction motifs and sequence covariation patterns.
Single-pass membrane receptor oligomers such as tyrosine kinase perform important cellular functions and are critically involved in diseases. However, their structure remains poorly characterized, which prevent the development of effective therapeutics. We developed a new technique to model single-pass membrane receptor oligomers guided by structural contacts solely predicted from genomic sequence covariations (Wang & Barth, Nat Comm 2015). Our technique enables unprecedented near-atomic accuracy prediction of a wide diversity of self-associated single pass transmembrane receptors and sets the stage for the efficient prediction of disease-related mutational effects on receptor structure and function.
We performed a structural bioinformatics analysis of the entire membrane proteome with known structures. We found that membrane proteins can be deconstructed in minimal interacting transmembrane helical trimers that adopt only 6 major topologies. For each topology, we identified consensus evolutionary conserved sequence motifs stabilizing the trimer conformations. Using these motifs, we were able to predict the topology of membrane protein domains and the local conformational flexibility of many multi-pass membrane proteins from sequence only. For the first time, our study revealed that common sequence-structure motifs forming consensus three-dimensional contacts govern the complex structure and plasticity of functionally unrelated multipass membrane proteins (Feng & Barth, Nat Chem Biol 2016). These findings pave the road for further understanding membrane protein folding and guiding the design of membrane proteins with novel structures and functions.
Genetic variations
3. Prediction of the effects of genetic variations on protein structure/function for precision personalized medicine.
Increasingly available data from whole human genome sequencing promotes new approaches using personal genome information to help diagnose, treat, and even prevent human diseases for which genetic variations can be risk factors or causative. However, the prediction of the pathogenicity of individual variations and the mechanistic relationships between pathogenic variations and their physiological consequences is partly limited by the lack of reliable predictions of their effects on the structure, dynamics, function, and interactions of specific proteins.
To address this limitation, we apply our efficient physical model of membrane protein structure and allostery (see protein design section) to predict mutational effects receptor stability, folding and signaling. We focus on the large family of GPCRs and investigate GPCR classes that have recently been found implicated in the development and progression of a wide array of cancers and cancer types. So far, studies have limited their scope to only a few receptors and a single cancer type. To gain a more holistic understanding of GPCR involvement in cancer, we will combine sequence bioinformatics and high-throughput structure modeling approaches to predict the structure and signaling effects of clinical mutations from The Cancer Genome Atlas (TCGA) across > 250 receptors. We hypothesize that, given a sufficiently large data set, we will identify conserved clusters of mutations within the GPCR structure fold with similar effects on receptor stability and signaling as well as correlations between mutations and specific signaling pathways or conserved functions in cancer progression. These studies will shed light on common mechanisms of cancer progression, and provide a rational basis for future personalized cancer diagnoses, risk stratifications and treatments.