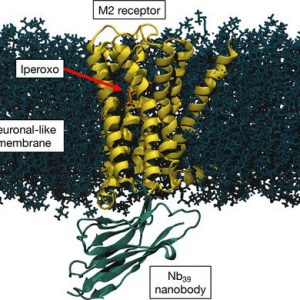
We simulate biological and biomimetic systems to uncover new insights about their structures, properties and interactions. We primarily integrate quantum mechanics/molecular mechanics (QM/MM) techniques with classical molecular dynamics and enhanced sampling methods. Additionally, we utilize machine learning techniques to bridge the gap between protein design and high level quantum mechanics to design proteins that bind cofactors with complex electronic structure such as zinc.
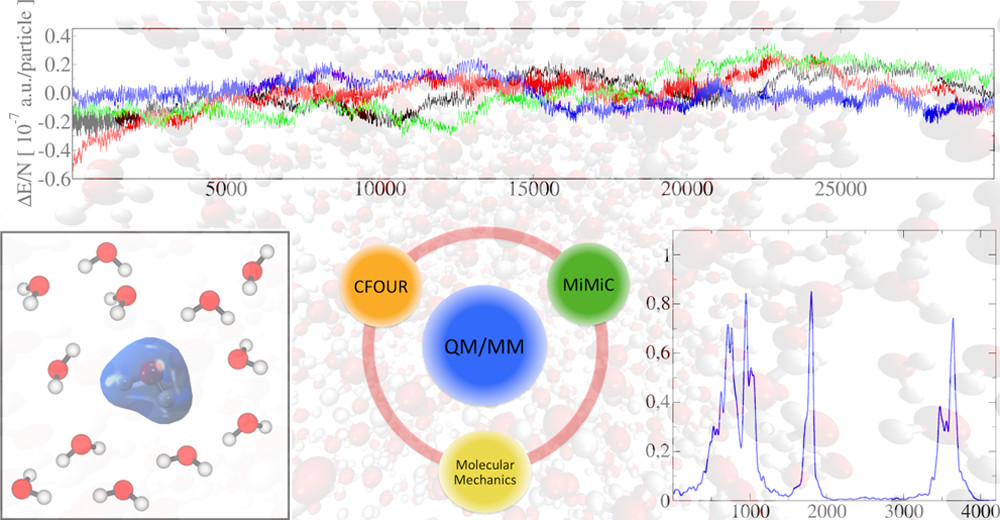
Ab initio molecular dynamics (MD) simulation based on density functional theory is a powerful tool in the study of physical and chemical systems. However, due to the larger size of realistic biological systems, a full ab initio treatment of the entire system is not feasible. Furthermore, such treatment is also not necessary as the chemically-relevant portion of a biological system is often limited in space and number of atoms. We cannot ignore the full system though, as the region of interest is still influenced by the biological environment.
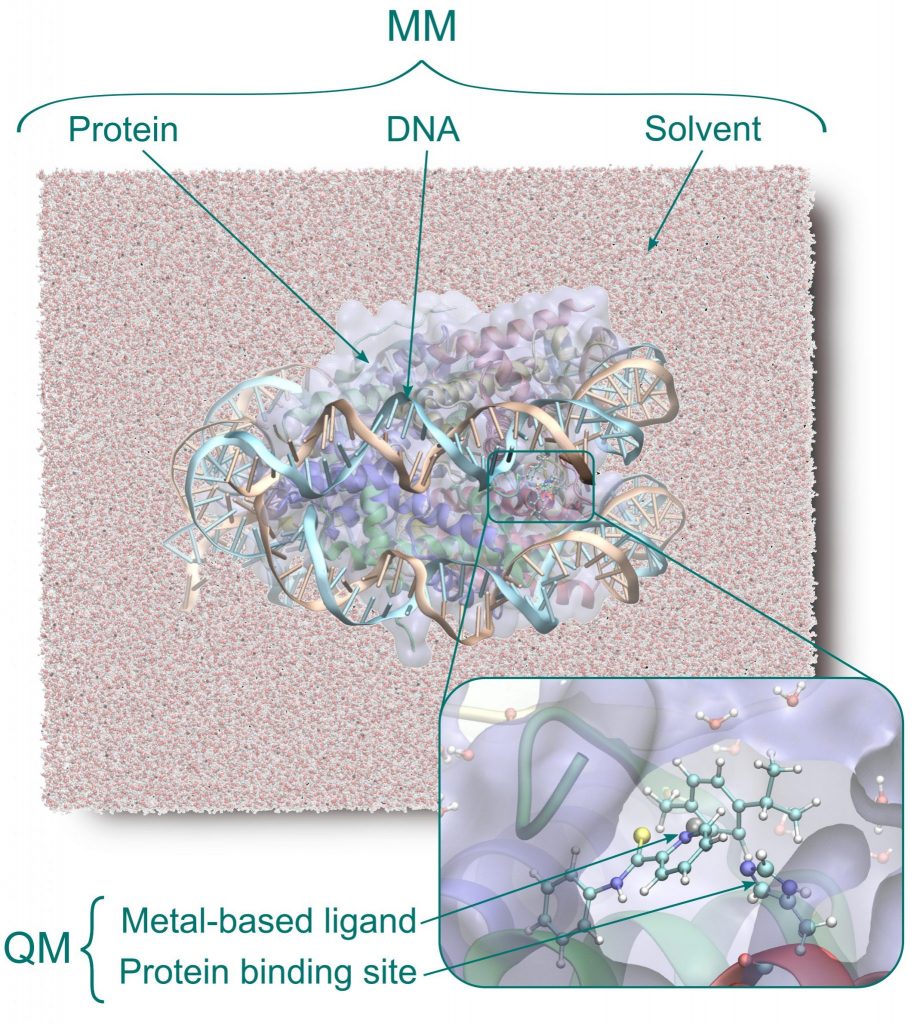
Quantum mechanics/molecular mechanics (QM/MM) approach allows for simulations of realistic biological systems by applying a classical (force field level) description whenever possible and a more accurate, but computationally more demanding, ab initio description where necessary, such as in the active site of an enzyme.
Our group integrates this hybrid QM/MM approach with other computational techniques, such as classical MD and enhanced sampling methods, to investigate mechanisms of biological interest. Current projects under study include:
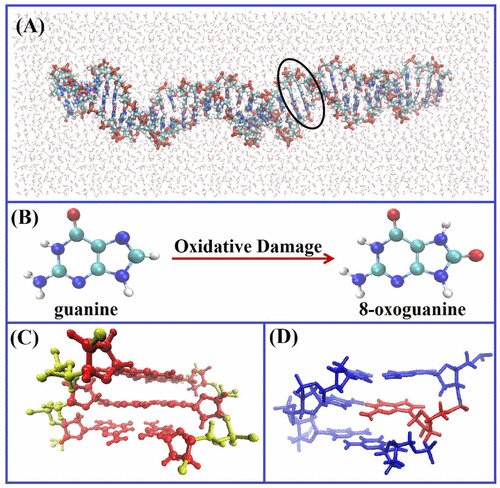
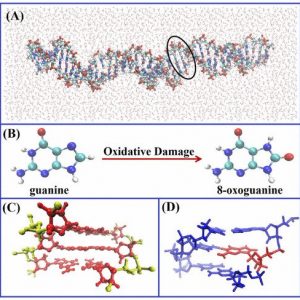
- DNA damage formation and repair
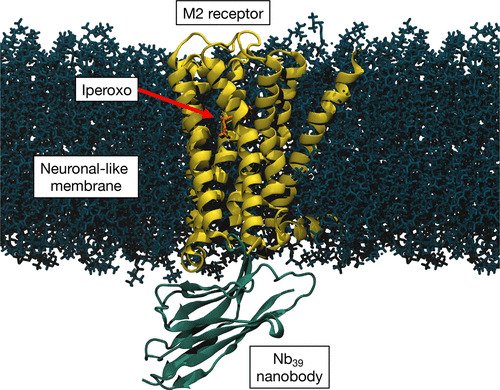
- Metal-based anticancer drugs
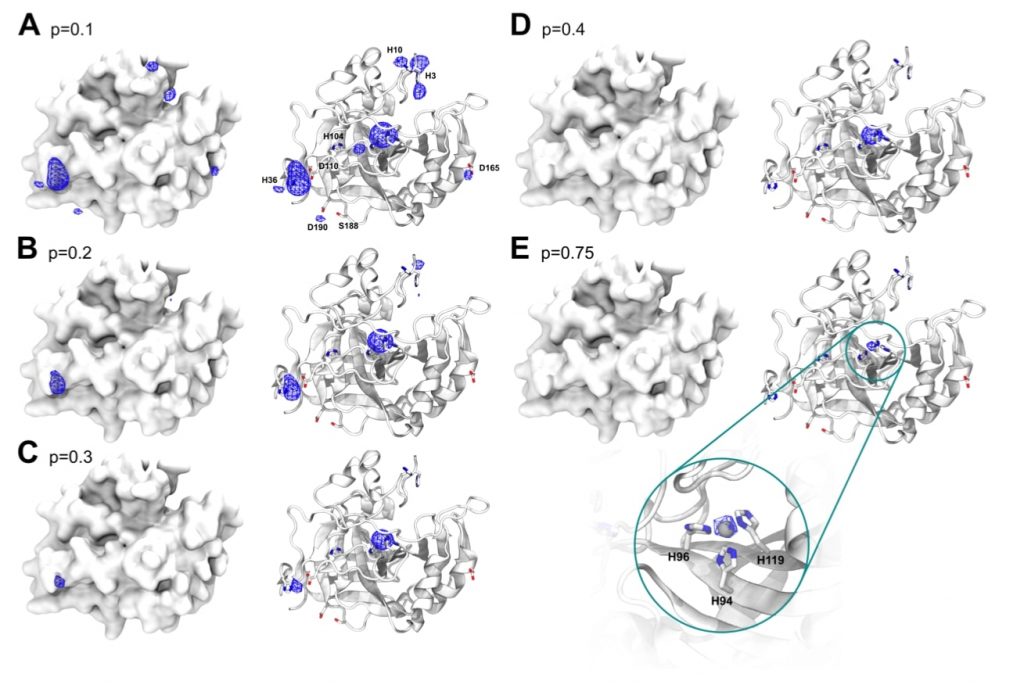
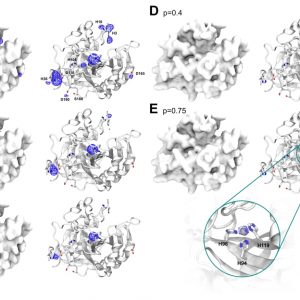
- Metalloprotein design