D. Ongari, L. Talirz, K. M. Jablonka, D. W. Siderius, and B. Smit, Data-Driven Matching of Experimental Crystal Structures and Gas Adsorption Isotherms of Metal–Organic Frameworks J. Chem. Eng. Data (2022) doi: 10.1021/acs.jced.1c00958
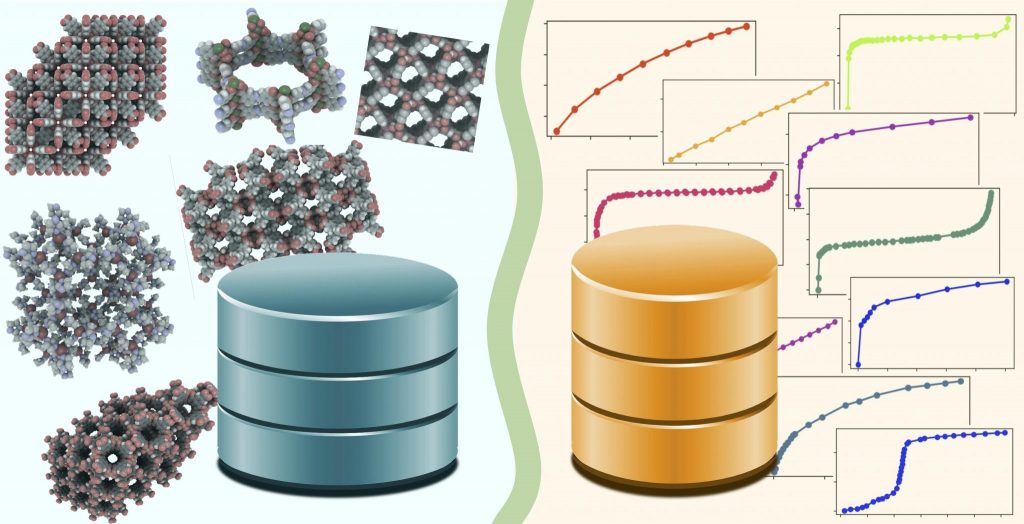
abstract: Porous metal–organic frameworks are a class of materials with great promise in gas separation and gas storage applications. Due to the high dimensional space of materials science and engineering, computational screening techniques have long been an important part of the scientific toolbox. However, a broad validation of molecular simulations in these materials is impeded by the lack of a connection between databases of gas adsorption experiments and databases of the atomic crystal structure of corresponding materials. This work aims to connect the gas adsorption isotherms of metal–organic frameworks collected in the NIST/ARPA-E Database of Novel and Emerging Adsorbent Materials to the corresponding crystal structures in the Cambridge Structural Database. With tens of thousands of isotherms and crystal structures reported to date, an automatic approach is needed to establish this link, which we describe in this paper. As a first application and consistency check, we compare the pore volume measured from low-temperature argon or nitrogen isotherms to the geometrical pore volume computed from the crystal structure. Overall, 545 argon or nitrogen isotherms could be matched to a corresponding crystal structure. We find that the pore volume computed via the two complementary methods shows acceptable agreement only in about 35% of these cases. We provide the subset of isotherms measured on these materials as a seed for a future and more complete reference data set for computational studies.