A. Ortega-Guerrero, M. Fumanal, G. Capano, and B. Smit, From Isolated Porphyrin Ligands to Periodic Al-PMOF: A Comparative Study of the Optical Properties using DFT/TDDFT J Phys Chem C (2020) doi:10.1021/acs.jpcc.0c06885
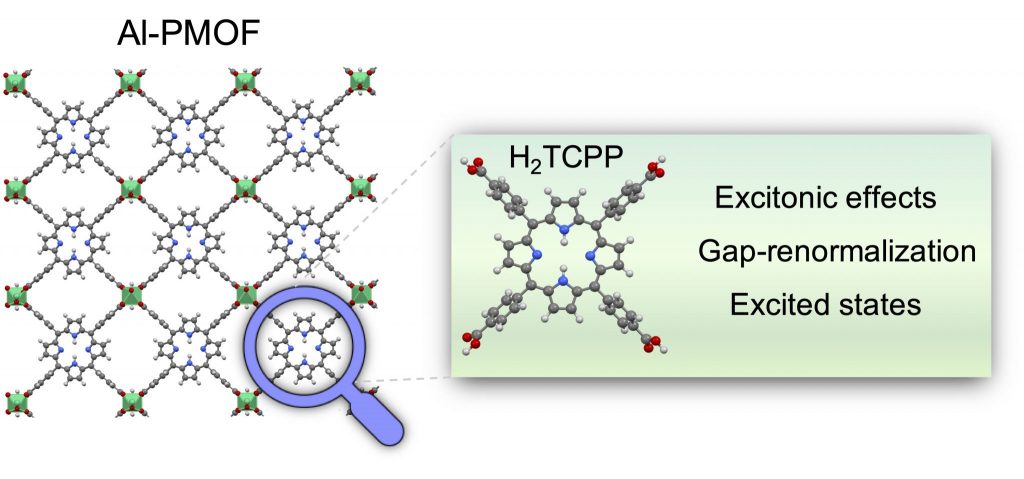
Abstract: The use of photosensitizers as organic ligands in Metal-Organic Frameworks (MOFs) is a common practice to engineer their UV/Vis optical absorption. For instance, MOFs consisting of porphyrin ligands usually inherit their light-harvesting properties and thus follow the Gouterman model in which the low-lying excitations correspond to π to π* transitions. However, the characterization of the excited states of porphyrin ligands in MOFs requires an appropriate description of the periodic crystal including the metal nodes and inter-molecular interactions. Here, we investigate the UV/Vis absorption properties of the porphyrin-MOF Al-PMOF and two metallated forms, Zn-Al-PMOF and Co-Al-PMOF, and of their corresponding isolated porphyrin ligands using DFT/TDDFT simulations with PBE, PBE0, and CAM-B3LYP functionals. Our results indicate that hybrid functionals are necessary to capture the proper nature of the transitions and the excitonic effects of the optical and fundamental gaps in porphyrin molecules and porphyrin-MOFs that PBE functional fails to describe. Likewise, the simulations show that a wrong representation of some excitations can be obtained depending on the functional and when the Tamm-Dancoff approximation is used. Finally, our results show that PBE and PBE0 functionals are not able to capture the gap-renormalization when going from the isolated molecules to the periodic crystals. Overall, the nature of the optical transitions, the excitonic effects, and the gap-renormalization are important features to assess in the prediction of optical properties in MOF crystals that require considering proper functionals and approximations to overcome the main failures of DFT/TDDFT calculations.