
ISIC’s Mass Spectrometry facility is equipped with different technologies covering a wide range of applications, from the analysis of small organic molecules to large biomolecules and metals. The high quality of mass spectrometry analyses can be achieved thanks to the various ionization techniques (ESI, APCI, APPI, CSI, MALDI, EI, CI, and ICP), mass analyzers (Quadrupole, Ion Trap, TOFs and Orbitraps) and separation techniques. Our facility also acquired a strong expertise in Top-Down Proteomics as well as Native MS for protein complexes analysis.
We provide service and scientific support to ISIC and EPFL research groups, external academics as well as external clients from industry (if desired under NDA).
Users can access the facility through instrument training and open access (contact us for training) or through sample submission in service mode as well as collaboration.
Beyond machine training, students can enroll in our PhD course given by the platform within the EDCH program of the Doctoral School of EPFL. You will find more information in the respective section on education.
If you have any questions, please drop us an email at: [email protected]
Main types of MS analyzes provided:
- High Resolution & high mass accuracy MS
- MALDI-TOF MS
- GC-MS
- Quantitative MS
- Protein intact mass denaturing & native MS
- Top/middle-down Proteomics (TDP)
- ICP-MS
High-resolution <5ppm mass accuracy MS
We perform HRMS spectra of ANY type of compound using different sample introduction techniques (direct injection or LC-MS), ionization techniques, and mass analyzers (TOF or Orbitrap). Main techniques are listed below:
 |

|
MALDI-TOF MS
We provide MALDI-TOF mass spectra of ANY type of compound using different sample preparation protocols (DD, Sandwich, Solvent-free, matrices, solvents,..) and modes of analysis (Reflectron & linear, polarity,..) depending on the type of compound. This technique is well suited for proteins, peptides, polymers and nanoparticles.

.
- Sample preparation for MALDI-MS:
-
For polymer samples, provide powder or in solution at concentrations of at least 1 mg/mL.
-
For polymers that cannot be dissolved in any solvent, solvent free deposition techniques can be used
-
Samples should not contain salts. Desalting can be performed prior to MALDI analyses by SPE (Sep-Pak, Zip-Tips), ultrafiltration or precipitation
-
The following buffers should be avoided:
– Detergents: Triton, SDS, Tween20
– Phosphate Buffered Saline , Sodium phosphate, Tris-HC, HEPES, MOPS
– Sodium bicarbonate
– Cell culture medium: CD-CHO, Opti-MEM I
– Glycerol
-
- MALDI recipies Database
- Use our Polymer MS Tool
GC-MS
We provide GC-MS analyses of volatile/semi-volatile compounds and mixtures of compounds, using either EI or CI ionization (NH3 as gas). In addition, we can perform Head-space GC-MS analyzes using our TriPlus RSH injector as well as manual gaz injections.
 |
 |
Quantitative MS
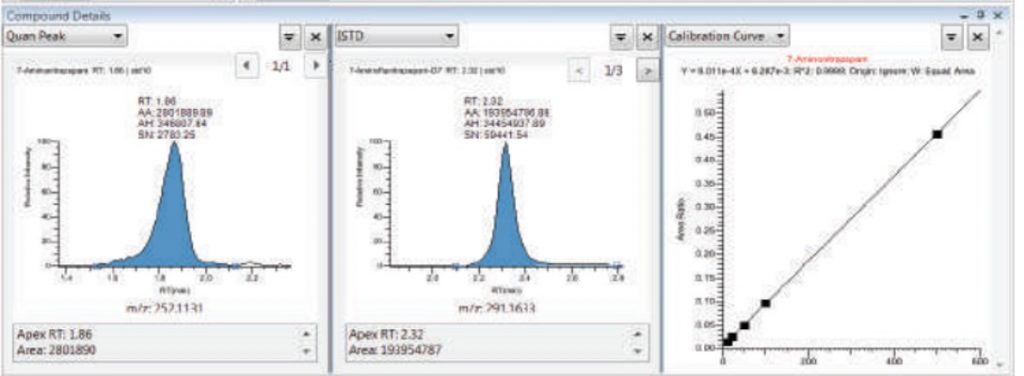
- LC-HRMS: we perform quantitation of drugs or in principle any small organic molecules in different matrices. Different LC stationary phases can be used and method development is generally required. The mass analyzer mostly used is a TOF. Quantitation is done using external calibration curves with freshly prepared samples. Adding an internal standard (ISTD) is the samples is better for improved robustness.
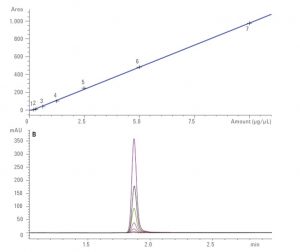
.
- GC-MS/MS: we perform quantitation of (semi)volatile compounds in different matrices. Sample preparation involves LLE and non-volatile samples should be derivatized (silylation) prior to analyses. Quantitation is done using external calibration curves with freshly prepared samples. Adding an internal standard (ISTD) is better for improved robustness.
Intact mass protein analysis
Intact mass protein analyses are performed using TOF or Orbitrap mass analyzers. Proteins up to 300 kDa can be in principle analyzed. Ionization techniques include ESI, MALDI and CSI. This last techniques allows the analysis of fragile protein complexes that are prone to degradation under standard heating ESI measurments.
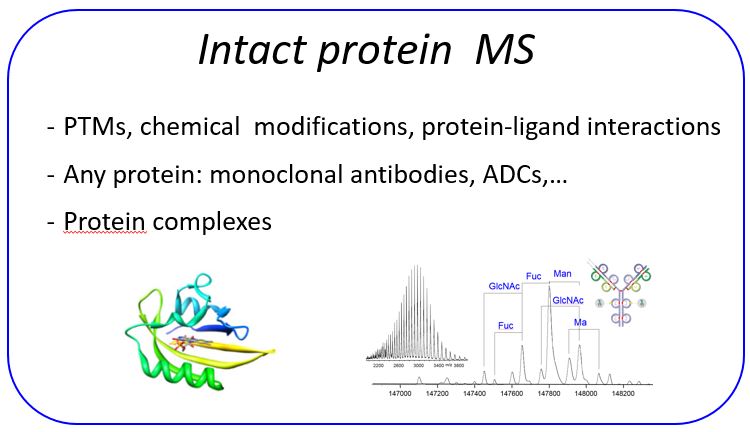
- Proteins are desalted using offline or online techniques depending on the protein purity and the amount available
- Intact mass measurments can be performed in denaturing and in native conditions:
Denaturing conditions
|
For unstructured proteins. Protein complexes are not preserved. Only covalent interactions are preserved
|
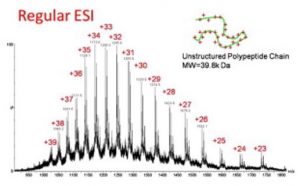 |
.Native conditions
|
Protein complexes are preserved = information on non covalent stoichiometry and structure of the complexes
|
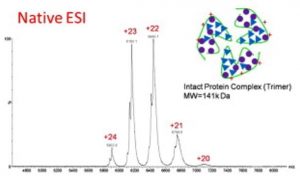 |
Middle/Down & Top-Down Proteomic-based analysis
For in-depth characterization of target proteins we provide middle-up/middle-down and top-down MS analysis using LC-MS/MS on an Orbitrap mass analyzer. Fragmentation of intact proteins or partially digested proteins allows identification and localization of PTMs/ other covalent modifications/amino acid substitutions within protein sequence. It is also a complementary method to classical bottom-up approach for protein identity confirmation. For complex protein mixtures top-down proteomic analysis can be performed in an untargeted manner.
For mAbs and ADC characterization in particular, we perform the following analyses:
- Intact mass analysis both in denaturing and native conditions
- TCEP reduction followed by middle-up analysis
- Enzymatic reaction with IdeS followed by middle-up analysis
- Enzymatic reaction with IdeS/TCEP reduction followed by middle-down analysis
- Multiplexing analysis is available for highly modified/complex samples
Workflow:
|
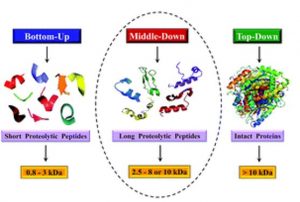 |
ICP-MS
We provide ICP-MS analysis for detection of metals (up to 33 elements simultaneously) in various matrices: metal alloys, aqueous solutions, nanoparticles, metal-organic compounds, biological samples, etc. Samples can be mineralized with microwave-assisted acidic digestion if needed (no HF digestion though). A multielement screening can be done in case of unknown composition of the sample, as well as a precise quantitation of selected elements. ICP-MS is a sensitive technique suitable for trace analysis where low detection limits (0.0005-1.0 ppb) are required.
